The Little Compound That Could: How Phenytoin Changed Drug Discovery and Development
On November 30, 1936, Tracy Putnam, Chief of Neurology at Boston City Hospital, wrote a letter to his friend, Oliver Kamm, Scientific Director at Parke, Davis and Company in Detroit. Putnam and his collaborator, H. Houston Merritt, had used a laboratory screening procedure to assess anticonvulsant activity, and one of the first Parke-Davis compounds they tested appeared particularly interesting (1).
The little molecule had a big name: 5,5-diphenylimidazolidine-2,4-dione. Because it consisted of two phenyl groups attached to a hydantoin ring, the investigators referred to the compound as diphenylhydantoin. In the 1970s, the generic name would be shortened to phenytoin.
When Heinrich Biltz first synthesized the compound in 1908 at the University of Kiel in Germany, no one predicted that it would become one of the most widely studied drugs of the twentieth century. Initially, it generated little interest. Biltz, an inorganic chemist, preferred to study auto-oxidation and oxidative degradation. Diphenylhydantoin was not synthesized again until the 1920s, when Arthur Dox, a chemist at Parke-Davis, prepared nineteen phenobarbital analogs in a search for hypnotic (i.e., sleep-inducing) drugs. Diphenylhydantoin, it turned out, was not a hypnotic, and Dox shelved the “inactive” compound (1–4).
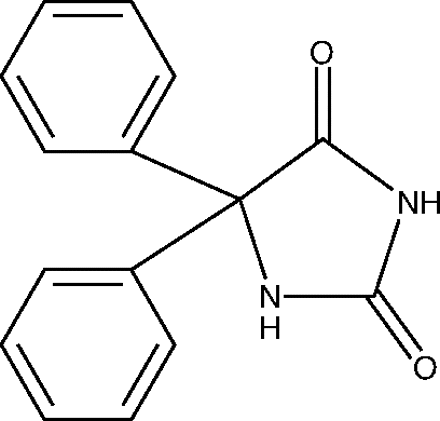
Phenytoin
About the same time, the dean at Harvard Medical School was taking steps to establish a department of neurology with a full-time faculty. Until then, most medical educators had depended on private practice for their income, limiting their ability to teach and do research. Stanley Cobb, a young neurologist from a prominent Boston family, was appointed as the salaried director of the new neurology group at Boston City Hospital, where the Harvard dean had already established a department of medicine (5).
The Boston City Hospital Team
Cobb was interested in epilepsy, and he attracted a team of like-minded researchers who would make quantum advances in the field, both experimentally and clinically. In addition to Putnam and Merritt, the talented group included William Lennox, Albert Grass, Erna Leonhardt, and Frederic Gibbs.
Lennox joined Cobb after returning from medical missionary work in China. His studies led him to differentiate three types of seizures—grand mal, petit mal, and psychomotor (now renamed generalized tonic-clonic, absence, and complex partial seizures, respectively). A life-long humanitarian, Lennox also established the Seizure Unit at Boston Children’s Hospital, which became the model for today’s comprehensive epilepsy centers (5). For his legendary contributions, a severe form of childhood epilepsy that he studied and characterized would be named in his honor (along with that of neurologist Henri Gastaut): the Lennox-Gastaut syndrome.
Lennox hired Erna Leonhardt initially as a technician, and she quickly became an integral member of Cobb’s team. Frederic Gibbs joined Cobb’s group after graduating from the Johns Hopkins Medical School. Fred and Erna soon married and thereafter they worked together, using the newly discovered electroencephalographic (EEG) technique in many pioneering studies of absence seizures and temporal lobe epilepsy (5, 6).
Albert Grass graduated from the Massachusetts Institute of Technology and then continued working at MIT as an electrical engineer. When Dr. and Mrs. Gibbs wanted to improve their single-channel EEG recorder to collect multichannel EEG data, Fred sought advice from MIT and hired Grass, who assembled the new instrument in his father’s basement. Grass and Gibbs continued to collaborate, making further improvements in the EEG and customizing other laboratory equipment, including most significantly, a stimulator. Eventually, Grass established the Grass Instrument Company in nearby Quincy, MA, manufacturing commercial polygraphs and other instruments for neurophysiologists (5, 6).
Tracy J. Putnam was a Boston Brahmin and used his prowess and connections to quickly build his reputation. He earned a Croix de Guerre for service as an ambulance driver during World War I. After graduating from Harvard Medical School, Putnam trained in neurosurgery. He spent a year in Europe conducting neurological research and then returned to Boston to became a neurosurgical resident under Harvey Cushing; he also trained in neurology under Cobb. At the time Cobb became head of the neurology unit at Boston City Hospital, Putnam formally held a research position in the hospital’s neurosurgery division, but he was an established researcher in both neurology and neurosurgery (5, 7). Putnam was more at ease in large rather than small interpersonal settings, more brilliant than practical, and more morally virtuous than politically correct (7, 8).

Tracy Putnam
H. Houston Merritt, a native of North Carolina, graduated from the Johns Hopkins Medical School, interned at Yale, and became a neurology resident at Boston City Hospital. After his residency and a “working” honeymoon trip to Europe, Merritt joined the medical faculty at Harvard and Cobb’s neurological research unit. He was an active laboratory researcher, conducting early studies on the constituents of cerebrospinal fluid. Merritt also quickly developed a reputation as a prodigious and insightful clinician who relished solving difficult clinical problems (5, 7, 8). An unassuming and modest man, he was also politically astute (8).

H. Houston Merritt
In 1934, Cobb left Boston City Hospital to take a position at Massachusetts General Hospital, and Putnam, who was then forty, immediately replaced him as head of the neurology unit. Merritt was thirty-two.
The Systematic Search for Anticonvulsant Drugs
Louis Pasteur said, “In the field of observation, chance favors the prepared mind.” Certainly, Putnam had inherited a thoroughly prepared group at Boston City Hospital. They were soon to make a “chance” observation that triggered a new epoch in the field of epilepsy (4, 9).
Although few drugs (e.g., bromide salts and phenobarbital) were available to treat epileptic patients in 1934, evidence was accumulating—primarily from metabolic studies—that seizures could be controlled by medicinal, rather than surgical, intervention. Researchers had demonstrated that hyperventilation induced petit mal attacks, which could be stopped by rebreathing into a paper bag and increasing the concentration of blood carbon dioxide (5). Lennox in particular was studying the effect of blood acidosis on seizures. Prolonged fasting produced acidic ketone bodies in the blood (the products of fat metabolism) and attenuated seizures. One treatment being investigated for epilepsy was a ketogenic diet that maintained the ratio of grams of fat to grams of protein plus carbohydrate above 3:1 (4, 10).
Meanwhile, the Gibbs couple had discovered that during a seizure, epilepsy patients exhibited excessive EEG discharges. Putnam reasoned that if an electrical brain storm characterized seizures, the best way to reliably trigger an experimental seizure was with an electric shock (5, 11). Drawing on recent reports by Spiegel and others who had developed similar instruments, Putnam worked with Gibbs and Grass to assemble a crude stimulator using parts from the electric motor of a salvaged WW I German airplane (5, 11–13).
Putnam and Merritt first used their stimulator to evaluate bromide and phenobarbital as positive controls. After cats were given sodium bromide (2 g), it took 50% more electrical current to generate a seizure than before; phenobarbital (0.1 g) increased the convulsive threshold three- to four-fold (14). Interestingly, they titrated each drug to a dose that was just sufficient to prevent cats from walking, prior to evaluation as an anti-convulsive agent. Most neurologists at the time thought that drugs suppressed seizures by a generalized sedative effect, but Putnam and Merritt’s results suggested the hypnotic and anticonvulsant effects could be separated.
In May, 1936, Putnam attended a presentation by Tinsley Harrison at the American Society for Clinical Investigation. Harrison, author of the widely used textbook of internal medicine and a professional acquaintance of both Putnam and Merritt, reported that kidney-failure patients with high blood concentrations of conjugated phenols did not exhibit convulsions, which otherwise normally occurred during uremia (5, 7, 15). This, and the fact that phenobarbital was more effective than most other barbiturates as an anticonvulsant, sparked Putnam’s interest in phenyl derivatives (14, 16, 17). He combed the catalogs of the Eastman Chemical Company and other suppliers for suitable phenyl compounds that were not obviously poisonous (5, 11).
Concurrently, Putnam had been contacting pharmaceutical houses, including Parke-Davis, requesting samples of barbiturate analogs and standard hypnotics. Only Arthur Dox, the Parke-Davis chemist, responded to his request. Dox had seven analogs available and shipped the lot to Boston on April 24, 1936 (1).
In the spring of 1936, Merritt and Putnam were honing their electrical stimulation technique to establish a standard procedure for assessing anticonvulsant activity. They settled on delivering a pulsed, square-wave current (80 per second) to cats through scalp-mouth electrodes for 10 seconds (Figure 1). At five minute intervals, they progressively increased the current until the animal exhibited a clonic-tonic seizure; this current was recorded as the animal’s control convulsive threshold (14, 17, 18). A test drug was then administered to the same animal and after 2–3 hours current was again applied in steps until convulsions occurred. A rating scale was used to group the drugs, based on the degree of elevation in convulsive threshold: 0, no change; +, 5 to 15 mA increase; 2+, 20 to 30 mA increase; 3+, 50 mA increase; and 4+, >50 mA increase (18).
Discovery of Phenytoin
In November, 1936, Putnam informed Parke-Davis of their initial results. The first drug on Dox’s list, the one he had shelved more than a decade earlier because it was not hypnotic, had impressive anticonvulsant activity (1). Diphenylhydantoin was now on the fast track.
Putnam and Merritt’s first paper appeared in Science in May, 1937. They reported that diphenylhydantoin, along with two other phenolic compounds, acetophenone and benzophenone, produced the greatest separation between anticonvulsant efficacy and hypnotic activity (14). On June 5, 1937, Merritt presented more complete results from their animal studies at the American Neurological Association meeting in Atlantic City (16).
Known neurodepressants such as alcohol and sodium pentobarbital raised the electrically induced convulsive threshold, but only at narcotic doses. A large group of known pharmacologic agents including aspirin, atropine, naphthalene, physostigmine, and pilocarpine were ineffective. Diphenylhydantoin was the most potent, nonsedating anticonvulsant (16, 17).
Because Dox had retired in September, 1936, W. Glen Bywater became the Parke-Davis chemist charged with selecting and sending additional compounds to Boston, and he communicated primarily with Merritt (1). Merritt and Putnam, with laboratory technician Dorothy Schwab, would eventually test more than 700 compounds in their screening model (Figure 1) (4, 18).
But their interest focused on diphenylhydantoin, the most effective compound from their early studies. Although regulators did not require preclinical safety testing (the Food, Drug and Cosmetic Act was signed in 1938), Otto Krayer (see 19), who had recently arrived to head Harvard’s pharmacology department and had credentials in toxicology, evaluated the drug in his laboratory. So did Oswald Gruhzit, the head of pharmacology and pathology at Parke-Davis (7, 20, 21). Cats, dogs, and rats tolerated high single and repeated doses of diphenylhydantoin without apparent untoward effects (20).
Clinical Efficacy of Phenytoin
In parallel, Parke-Davis prepared diphenylhydantoin as the sodium salt, which was much more soluble than the free acid, and by January, 1937, sufficient supplies were available for clinical trials. Merritt started treating epilepsy patients with sodium diphenylhydantoinate in May, the same month the Science paper appeared. With results from the first eight patients, Putnam reported clinical efficacy to Parke-Davis in August, 1937, thus confirming their animal observations (1, 4) (Boxes 1 and 2).
Clinical Efficacy in Generalized Seizures
“Case 2. A. W., a white man, aged 26, suffered from severe grand mal attacks of five years’ duration, occurring almost daily with an increase in frequency to two or three attacks daily during the month before entry to the hospital, although the patient was receiving 6 grains (0.4 Gm.) of phenobarbital daily... Electroencephalography showed frequent bursts of abnormal cortical activity of the grand mal type. Treatment was started Dec. 2, 1937, with 0.1 Gm. of sodium diphenylhydantoinate three times a day. The patient has been entirely free of attacks since starting treatment” (20).
Clinical Efficacy in Complex Partial Seizures
“Case 5. S. B. R., a white man, aged 54, an insurance salesman, gave a history of attacks of five years’ duration, occurring two or three times a day, characterized by a turning of the head to one side, chewing movements of the jaw, mental cloudiness and performance of automatic actions, such as taking off shoes. The patient would not fall but would be unable to answer questions or talk coherently for from thirty to ninety seconds... Electroencephalography showed evidences of abnormal cortical activity typical of those found in patients with psychomotor equivalents. Previous treatment had consisted of 9 grains (0.6 Gm.) of phenobarbital and 30 grains (2 Gm.) of sodium bromide daily, plus calcium gluconate, with no effect on the severity or frequency of attacks. The patient was started on treatment with sodium diphenylhydantoinate Aug. 8, 1937, in doses of from 0.4 to 0.6 Gm. a day. The frequency of attacks has decreased from two or three attacks daily to an average of three attacks a month” (20).
Subsequently, Parke-Davis sent clinical supplies to O. P. Kimball, who began treating children with sodium diphenylhydantoinate at the Detroit Epilepsy Clinic in January, 1938, and to J. L. Fetterman of Cleveland’s Lakeside Hospital epilepsy clinic and Western Reserve School of Medicine in May, 1938 (7, 22).
Merritt and Putnam first publically disclosed the drug’s clinical efficacy to the Section on Nervous and Mental Diseases of the American Medical Association on June 17, 1938 in San Francisco (5, 7, 20). By that time, they had already treated 200 adult and pediatric patients whose seizures were refractory to other anticonvulsant treatments. Sodium diphenylhydantoinate had eliminated or greatly reduced seizures in 85% of the 142 patients who had been treated for two to eleven months (20, 23).
On June 23, 1938, Parke, Davis and Company added sodium diphenylhydantoinate (Dilantin®) to its list of marketed products. (1) Merritt and Putnam’s clinical results appeared in the September 17, 1938, issue of the Journal of the American Medical Association, which at that time was mailed to nearly all American physicians (5, 20).
Merritt continued enrolling and treating patients with diphenylhydantoinate and reported updated results at the American Neurological Association and American Psychiatric Association meetings in 1939 (7, 23–25). He had now treated more than 350 previously refractory patients, including all of Lennox’s epilepsy patients. Sodium diphenylhydantoinate effectively controlled most patients with psychomotor or grand mal seizures, but only about half of the petit mal patients responded to the drug (23, 25). Merritt and Putnam’s results suggested that different types of seizures might respond differently to the same drug (7).
Later in 1939, the American Medical Association Council on Pharmacy and Chemistry accepted Dilantin® into its drug compendium, New and Nonofficial Remedies, and indicated the drug for epileptic patients who were refractory to phenobarbital or bromides and for those in whom those drugs produced disagreeable side effects. Of the thirteen clinical trials and 595 patients used in the council’s evaluation, Merritt and Putnam’s studies totaled 34% of the cases (4, 7).
The Influence of Phenytoin
Phenytoin’s popularity grew quickly, and as early as 1940 it was hailed as initiating a new epoch in the treatment of epilepsy (9). Merritt and Putnam’s findings motivated researchers to seek even more effective drugs, and pharmaceutical companies set up aggressive screening programs. In the next two decades, a dozen new anticonvulsants were introduced into clinical therapy (5, 7). Drug screening then waned until the 1970s, when J. Kiffin Penry, concerned about a still sizable population of intractable patients, rekindled efforts to find new and more effective drugs (26, 27). But phenytoin remained a first-line treatment until the 1980s (4).
In addition, phenytoin generated an enormous literature, appearing in more than 15,000 research papers. Its nonlinear pharmacokinetics, highly variable absorption, P450-mediated metabolism, broad therapeutic utility, and many interactions with other drugs made phenytoin a popular research topic for pharmacologists of every stripe and the subject of numerous dissertations (4).
Phenytoin’s mechanism(s) of action likewise intrigued and baffled researchers for many years. Eventually, McLean and Macdonald showed that phenytoin produced a use-dependent blockade of the neuronal sodium channel at clinically relevant anticonvulsant concentrations (28). The drug’s ability to stabilize excitable membranes seems to account not only for its anticonvulsant action but also its usefulness in certain cardiac arrhythmias (29).
Phenytoin’s pharmacologic profile includes many potential side effects. In their earliest clinical reports, Merritt and Putnam noted the drug’s now familiar actions: skin rash, ataxia, hirsutism, nystagmus, and gum hyperplasia (20, 23, 24). Other undesirable features (e.g., osteomalacia, peripheral neuropathy, and immunologic effects) were discovered more slowly (4). Teratogenicity and the fetal hydantoin syndrome also were first noted only after decades of use (30–32).
Despite all its faults, phenytoin remains one of the most widely used drugs in the world. Although in most cases it is now a second-line therapy, phenytoin is still the drug of choice in the emergency treatment of seizures and in status epilepticus (4).
Merritt and Putnam, After Phenytoin
As for Merritt and Putnam, their last co-authored papers appeared in 1945, ten years after they began their quest for a new anticonvulsant. Phenytoin had unleashed widespread interest in anticonvulsant research, and to aid those investigators they published the complete list of compounds they had tested, along with summary results (18). Parke-Davis chemists had synthesized the majority of those compounds, many of which were hydantoin analogs, and Merritt and Putnam collaborated with Bywater to explore hydantoin structure-activity relationships (33) (Table 1). Having already secured the rights to the molecule from Biltz, Parke-Davis received its first diphenylhydantoin patent in October, 1946 (5).
After announcing their success with phenytoin, Merritt and Putnam also tested samples submitted by Smith, Kline and French Laboratories, the William S. Merrell Company, Mallinckrodt Chemical Works, and a professor at the University of Texas (18). Of the sevety-six compounds that received a 4+ rating, Merritt evaluated five in clinical studies. However, phenytoin, their earliest discovery, proved superior to the other four and was the only commercial drug to emerge from their efforts (4).
Putnam had left Boston City Hospital in 1939 while the animal and clinical studies were still in progress to become director of the Neurological Institute of New York, a unit within the Columbia-Presbyterian Medical Center. His position gave him the titles of professor of neurology at Columbia Medical School and director of both the neurology and neurosurgery services at Presbyterian Hospital. Although he enhanced the struggling institute’s prestige despite distractions from war-related activities, Putnam’s relations with the hospital’s administration became increasingly strained. The dispute culminated with a leave of absence in 1946 and his resignation the following year. Putnam spent his remaining years in Los Angeles, where he served briefly as chief of neurosurgery at Cedars of Lebanon Hospital (5). His interests shifted to multiple sclerosis and surgery for movement disorders, but his bold ideas were no longer tempered by pragmatic colleagues. He tended to alienate unsympathetic peers, and his personality, reserved by nature, further isolated him (8). He saw fewer and fewer patients in his private practice, and phenytoin turned out to be the high point of his career. He died in 1975 at the age of eighty-one.
Merritt left Boston City Hospital in 1945 to head a newly created institute at Montefiore Hospital in New York. In 1948, he followed Putnam as head of the Neurological Institute at Columbia-Presbyterian. Building on the talent that Putnam had recruited, Merritt created a first-rate research and training program. At least forty-five of his residents later became chiefs of neurology at other medical schools. In addition to his longstanding interest in cerebrospinal fluid and epilepsy, Merritt also published books of his clinical research achievements in neurosyphilis, headache, stroke, multiple sclerosis, and genetic diseases (5). In 1958, he became dean of the College of Physicians and Surgeons of Columbia University, as well as vice president of health services (34). As a university administrator, Merritt was instrumental in attracting donations for a new twenty-story research building (5). In summing up Merritt’s precocious clinical acumen, absolute freedom from affection, unfailing good nature, and unerring diagnostic prowess, a contemporary remarked, “He was extremely well put together” (8). Merritt died in 1979 at the age of seventy-seven.
Merritt and Putnam’s Legacy
Although their paths diverged, one achieving greatness and the other fading from view, Merritt and Putnam’s work at Boston City Hospital represents a pharmacology milestone. They devised a simple and reliable method to assess drugs for anticonvulsant effects and then used it to systematically screen hundreds of compounds for efficacy. They showed that the anticonvulsant and sedative effects of drugs could be separated, inspiring not only their own research but also that of many other investigators for more selective drugs. They validated their laboratory observations by demonstrating that their animal model accurately predicted anticonvulsant efficacy in humans, and phenytoin, their first discovery, was also the first anticonvulsant drug to be tested in animals before it was given to humans. They then used phenytoin to show that a drug could have selective activity, effectively treating some seizure types better than others. This not only had therapeutic implications for the choice of drugs to treat individual patients, it also implied that different pathogenic mechanisms were responsible for different seizure types.
Their successful experimental paradigm became the blueprint that many drug companies adopted to discover and develop other anticonvulsant drugs, an approach now called translational research. The application of basic science results from the laboratory to a clinical application that provides an effective treatment in humans is now emerging as the preferred strategy, in general, for discovery and development of new drugs (35).
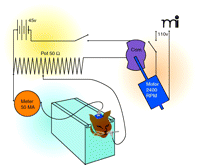
Apparatus used in(14). Reprinted with permission.
Anticonvulsant Activity of 5-alkoxymethyl and 5-phenoxymethyl-5-alkylhydantoins
Acknowledgments
The author wishes to thank Dr. Arthur Raines for his careful review and comments on the draft manuscript.
- Copyright © 2009
References

Rebecca J. Anderson, PhD, holds a BA in chemistry from Coe College and her doctorate in pharmacology from Georgetown University. After several industry positions in pharmacological research and development, she now works as a technical writer and is the 2009 recipient of the Larson Award from the American Medical Writers Association. E-mail rebeccanderson{at}msn.com

