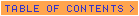|
|
Microscopic Polyangiitis in a Pediatric Patient
Ala'addin Kandeel, MD;
Sujatha Ramesh, MD;
Yongxin Chen, MD;
Cazibe Celik, MD;
Edwin Jenis, MD;
Julian L. Ambrus, Jr, MD
Arch Fam Med. 2000;9:1189-1192.
ABSTRACT
Microscopic polyangiitis (MPA), previously called hypersensitivity angiitis, is a systemic necrotizing vasculitis that involves many organ systems including the skin, joints, kidneys, and lungs. Microscopic polyangiitis most commonly affects adults in the fourth and fifth decades of life, with only a few cases reported in children. We describe a pediatric patient with microscopic polyangiitis.
REPORT OF A CASE
A 17-year-old male with no significant medical history was admitted to the hospital because of respiratory distress. He had an active high school social life with no history of sexual activity or substance abuse. Family history was notable for hypertension and lung cancer.
After returning from a trip to Florida, the patient noted lethargy, pain, and swelling in his feet, which was worse in the morning and improved with use. A month later he noted joint pain in his hands, shoulders, knees, and feet, without warmth or erythema. Scleral hyperemia was present each morning. He had lost 9 kg because of anorexia. At that time, Epstein-Barr virus and Lyme serology tests were negative. The complete blood cell count was normal. The patient was given ibuprofen. One month later he developed a cough with blood-tinged sputum, epigastric pain, diarrhea, and a temperature of 39°C. He was admitted to the hospital.
At the time of admission, the temperature was 37.2°C, the pulse was 100 beats per minute, and the respiratory rate was 25 breaths per minute. The blood pressure was 130/80 mm Hg. A nodular erythematous rash was noted on the elbows and lateral ankles. The conjunctivae were hyperemic, and a superficial ulcer was noted in the buccal mucosa. He had an area of necrosis in the left tonsil. A chest examination revealed bilateral inspiratory crackles. A heart examination revealed no gallop, but there was a grade 1 systolic murmur at the left sternal border. The abdomen was tender to palpation in the left lower quadrant with no rebound tenderness. A stool sample was positive for occult blood. No synovitis was present. Muscle strength and neurologic examination results were normal.
A laboratory examination revealed a white cell count of 6.9 x 109/L with 0.50 neutrophils, 0.20 bands, 0.13 lymphocytes, and 0.07 eosinophils. Hemoglobin level was 116 g/L, and platelet count was 213 x 109/L. Creatinine/serum urea nitrogen ratio was 0.9/9 mg/dL, aspartate aminotransferase/alanine aminotransferase ratio was 107/282 U/L, alkaline phosphatase level was 123 U/L, lactate dehydrogenase level was 217 U/L, total bilirubin level was 20.52 µmol/L (1.2 mg/dL), albumin level was 30 g/L, total protein level was 61 g/L, and electrolyte levels were normal. Erythrocyte sedimentation rate was 68 mm/h. Prothrombin time and partial thromboplastin time were normal. The urine revealed 2+ protein with moderate blood; the sediment contained 2 white cells per high-power field and 3 to 5 red cells per high-power field. The chest x-ray at admission showed bibasilar infiltrates and no cardiomegaly (Figure 1).
|
|
|
|
Figure 1. Admission chest x-ray showing bibasilar infiltrates.
|
|
|
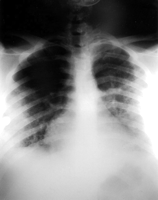
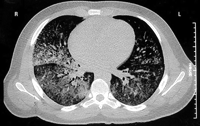
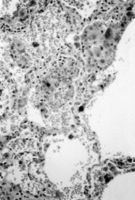
Treatment with intravenous gentamicin and clindamycin was begun. Within 24 hours, respiratory distress and hemoptysis worsened. The hemoglobin level dropped to 91g/L, and the patient was transferred to the intensive care unit for further management. Additional laboratory studies included quantitative immunoglobulins, which were at normal levels. Purified protein derivative (tuberculin) testing was negative, with positive controls. Complement levels were normal. Serologic testing for cytomegalovirus, Epstein-Barr virus, hepatitis C, respiratory syncytial virus, Mycoplasma, Legionella, leptospirosis, and human immunodeficiency virus was negative. Blood, urine, and throat cultures were negative for viral and bacterial organisms. A computed tomographic scan of the chest revealed bibasilar fluid (Figure 2). Cytoplasmic anitneutrophil cytoplasmic antibody (c-ANCA) titers were positive at 1:100, and tests for perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) and antiglomerular basement membrane antibodies were negative. Twenty-four-hour urine collection yielded 5.7 g of protein and a creatinine clearance of 1.97 mL/s (118 mL/min). A biopsy specimen of the skin rash showed neutrophilic vasculitis involving capillaries, venules, and arterioles (Figure 3). Intravenous methylprednisolone sodium succinate was given every 6 hours. Respiratory distress worsened. An open-lung biopsy was performed, and the specimen showed capillaritis and arteriolitis without granuloma formation (Figure 4).
|
|
|
|
Figure 2. Computed tomagraphic scan of the chest showing bibasilar fluid.
|
|
|
|
|
|
|
Figure 3. Biopsy of skin lesion showing a neutrophilic vasculitis involving capillaries, venules, and arterioles.
|
|
|
|
|
|
|
Figure 4. Biopsy of the lung showing capillaritis and arteriolitis.
|
|
|
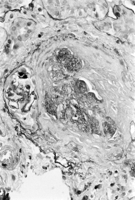
Daily low-dose cyclophosphamide therapy was instituted. Respiratory distress rapidly resolved, and the patient was discharged 6 days later while taking daily steroids and cyclophosphamide. He was completely asymptomatic after 4 weeks and returned to his normal energy and activities. A renal biopsy specimen, obtained months after discharge because of persistent hematuria and proteinuria, revealed pauci-immune crescentic glomerulonephritis (Figure 5).
|
|
|
|
Figure 5. Biopsy of the kidney showing crescentic glomerulonephritis. Immunofluorescence staining for IgG, IgA, and IgM was negative.
|
|
|
COMMENT
The patient had pulmonary renal syndrome and the characteristic manifestations of microscopic polyangiitis (MPA). Nongranulomatous vasculitis was demonstrated in the skin, lungs, and kidneys. A positive ANCA titer was noted, but there was no immune complex deposition in the kidneys. Interestingly, the c-ANCA titer was positive rather than the p-ANCA, which is more common in MPA.1
Microscopic polyangiitis is a systemic necrotizing nongranulomatous vasculitis that affects small blood vessels and is associated with focal segmental necrotizing glomerulonephritis. It was first recognized and distinguished from polyarteritis nodosa in the 1950s by its involvement of vessels smaller than arteries (ie, arterioles, venules, and capillaries).2-3 It was distinguished from Wegener granulomatosis in the 1980s based on the differences in lung abnormalities. The average age of onset is 50 years, and it is very rare in children. The disease affects both sexes equally. Clinical features include constitutional symptoms such as fever, anorexia, fatigue, and weight loss. Another characteristic feature is rapidly progressive glomerulonephritis, which often affects the kidneys in the early stages of the condition. Pulmonary hemorrhage is seen in 30% of MPA cases, but it is rarely found in Wegener granulomatosis and never in classic polyarteritis nodosa.4
Skin lesions include purpura and splinter hemorrhages, which occur in 50% of patients. Myalgias and arthralgias are present in up to 70% of patients, whereas frank arthritis is less common. Microscopic polyangiitis can affect the gastrointestinal tract in 30% of cases and result in abdominal pain, diarrhea, and bleeding. Peripheral neuropathy occurs in about a third of patients. Ocular lesions are seen in MPA and may include conjunctivitis, scleritis, and uveitis.5-7 Antineutrophil cytoplasmic antibodies, mainly p-ANCA, are present in 50% of patients.8 Biopsy of the lung or kidney is essential to demonstrate both small-vessel vasculitis and the absence of granuloma and of immune complex deposition, and to distinguish MPA from Wegener granulomatosis, Goodpasture syndrome, systemic lupus erythematosus, mixed cryoglobulinemia, Behçet disease, Henoch-Schonlein syndrome, and drug reactions.
The prognosis and outcome of MPA depends on the extent and severity of organ involvement.9 Early diagnosis and prompt institution of steroids and cyclophosphamide is essential.10 Because of the high relapse rate,11 treatment for up to 12 months is often needed. Azathioprine can be used to maintain remission in patients who develop hemorrhagic cystitis.
Our report describes MPA in an adolescent patient. Microscopic polyangiitis is rare in children but should be promptly diagnosed because it may cause mortality if left untreated.
AUTHOR INFORMATION
Accepted for publication September 13, 2000.
We wish to express our appreciation to Natalie Brock for her excellent secretarial assistance.
Corresponding author and reprints: Julian L. Ambrus, Jr, MD, Division of Allergy, Immunology, & Rheumatology, Department of Medicine, Buffalo General Hospital, State University of New York at Buffalo, 100 High St, Buffalo, NY 14203 (e-mail: jambrus{at}acsu.buffalo.edu).
From the Division of Allergy, Immunology, & Rheumatology, Department of Medicine (Drs Kandeel, Chen, and Ambrus), and the Department of Pathology (Dr Celik), Buffalo General Hospital; the Department of Pediatrics, Children's Hospital of Buffalo (Dr Ramesh); and the Department of Pathology, Millard Fillmore Hospital (Dr Jenis); State University of New York at Buffalo.
REFERENCES
| |
1. Hauschild S, Schmitt W, Csernok E. ANCA in systemic vasculitides, collagen vascular disorders and inflammatory bowel diseases. In: Gross WL, ed. ANCA-Associated Vasculitis. New York, NY: Plenum Press; 1993:245-254.
2. Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum. 1994;37:187-192.
ISI
| PUBMED
3. Guillevin L, Lhote F. Distinguishing polyarteritis nodosa from microscopic polyangiitis and implications for treatment. Curr Opin Rheumatol. 1995;7:20-24.
PUBMED
4. Jennings CA, King TE Jr, Tuder R, Cherniack RM, Schwarz MI. Diffuse alveolar hemorrhage with underlying isolated pauci-immune pulmonary capillaritis. Am J Respir Crit Care Med. 1997;155:1101-1109.
ABSTRACT
5. Lhote F, Cohen P, Genereau T, Gayraud M, Guillevin L. Microscopic polyangiitis: clinical aspects and treatment. Ann Med Interne (Paris). 1996;147:165-177.
PUBMED
6. Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med. 1985;56:467-483.
7. Zashin S, Fattor R, Fortin D. Microscopic polyarteritis: a forgotten aetiology of haemoptysis and rapidly progressive glomerulonephritis. Ann Rheum Dis. 1990;49:53-56.
FREE FULL TEXT
8. Gross WL, Csernok E. Immunodiagnostic and pathophysiologic aspects of anti-neutrophil cytoplasmic antibodies in vasculitis. Curr Opin Rheumatol. 1995;7:11-19.
PUBMED
9. Fuiano G, Cameron J, Raftery M, Hartley BH, Williams DG, Ogg CS. Improved prognosis of renal microscopic polyarteritis in recent years. Nephrol Dial Transplant. 1988;3:383-391.
FREE FULL TEXT
10. Fauci AS, Katz P, Haynes BF, Wolff SM. Cyclophosphamide therapy of severe systemic necrotizing vasculitis. N Engl J Med. 1979;301:235-238.
ABSTRACT
11. Gordon M, Luqmani RA, Adu D, et al. Relapses in patients with a systemic vasculitis. Q J Med. 1993;86:779-789.
|