
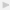

Data Integration in Genetics and Genomics: Methods and Challenges
- Jemila S. Hamid jemila{at}utstat.toronto.edu1
- Pingzhao Hu phu{at}sickkids.ca2
- Nicole M. Roslin nroslin{at}sickkids.ca2
- Vicki Ling vicki.ling{at}utoronto.ca1,3
- Celia M. T. Greenwood celia.greenwood{at}utoronto.ca4
- Joseph Beyene joseph{at}utstat.toronto.edu1,2,4
- 1Biostatistics Methodology Unit, The Hospital for Sick Children Research Institute, 555 University Avenue, Toronto, ON, Canada, M5G 1X8
- 2The Center for Applied Genomics, The Hospital for Sick Children Research Institute, 555 University Avenue, Toronto, ON, Canada, M5G 1X8
- 3Program in Developmental and Stem Cell Biology, The Hospital for Sick Children Research Institute, 555 University Avenue, Toronto, ON, Canada, M5G 1X8
- 4Dalla Lana School of Public Health, University of Toronto, 555 University Avenue, Toronto, ON, Canada, M5G 1X8
Abstract
Due to rapid technological advances, various types of genomic and proteomic data with different sizes, formats, and structures have become available. Among them are gene expression, single nucleotide polymorphism, copy number variation, and protein-protein/gene-gene interactions. Each of these distinct data types provides a different, partly independent and complementary, view of the whole genome. However, understanding functions of genes, proteins, and other aspects of the genome requires more information than provided by each of the datasets. Integrating data from different sources is, therefore, an important part of current research in genomics and proteomics. Data integration also plays important roles in combining clinical, environmental, and demographic data with high-throughput genomic data. Nevertheless, the concept of data integration is not well defined in the literature and it may mean different things to different researchers. In this paper, we first propose a conceptual framework for integrating genetic, genomic, and proteomic data. The framework captures fundamental aspects of data integration and is developed taking the key steps in genetic, genomic, and proteomic data fusion. Secondly, we provide a review of some of the most commonly used current methods and approaches for combining genomic data with focus on the statistical aspects.
1. Background
In recent years, increasing amounts of genomic data have become available. The size, type, and structure of these data have also been growing at an unprecedented rate. Gene expression, single nucleotide polymorphisms (SNP), copy number variation (CNV), proteomic, and protein-protein interactions are some examples of genomic and proteomic data produced using high throughput technologies such as microarrays [1], array comparative hybridization, aCGH [2], and mass spectrometry [3]. Each of these distinct data types provides a different, partly independent and complementary view of the whole genome. However, elucidation of gene function and other aspects of the genome may require more information than is provided by one type of data. The amount and type of biological data are expected to increase even further (e.g., methylation, alternative splicing, transcriptomic, metabolomic, etc.). This proliferation of experimental data makes systematic integration an important component of genomics and bioinformatics [4]. Data integration is increasingly becoming an essential tool to cope with the ever increasing amount of data, to cross-validate noisy data sets, and to gain broad interdisciplinary views of large genomic and proteomic data sets. Instances of combining and synthesizing data have increased considerably in the last several years and the need for improved and standardized methods has been recognized [5–7].
In functional genomics, for example, one is interested in defining the function of all the genes in the genome of an organism. Defining functions of genes is a daunting task and achieving this goal requires integrating information from different experiments [8]. Similarly, classifying a protein as a membrane protein based on protein sequences is a nontrivial task and has been the subject of much previous research, and it has been demonstrated that incorporating knowledge derived from aminoacid sequences, gene expression data, and known protein-protein interactions significantly improves classification performance compared to any single type of data [9].
The need for integration of heterogeneous data measured on the same individuals arises in a wide range of clinical applications as well. In this regard, the best example is perhaps the challenge that cancer researchers and clinicians face in the diagnosis, treatment, and prognostication of this complex disease. The clinical management of cancer is currently based for the most part on information accumulated from clinical studies [10]. However, cancer is thought to be primarily caused by random genetic alterations, and as such genomic data such as gene expression and protein data can be used to classify tumors into subtypes, and thus may have the potential to improve the clinical management of cancer.
Data integration also plays an important role in understanding environment-genome interactions in toxicogenomics, a discipline where one investigates how various genes respond to environmental toxins and stressors, and how these factors modify the function and expression of genes in the genome [11]. The contribution of different sources of data (such as genomics, proteomics, and SNP) in advancing the field of toxicology is discussed by Patel et al. [11].
Another more common type of data integration relates to combining similar types of data across different studies. This can, for example, be done through meta-analytic approaches. For instance, with the increasing number of publicly available independent microarray datasets, it is important to combine studies that address similar hypotheses. Such analyses have proven to be very useful in several applications (see, e.g., Rhodes et al. [12]).
There are a number of challenges in the context of high biology genetic and genomic studies. The challenges may be of conceptual, methodological, or practical nature and may relate to issues that arise due to experimental, computational, or statistical complexities. For example, genomic data are often subject to varying degrees of noise, the curse of high dimensionality, and small sample sizes. One may, therefore, benefit from integrating clinical, genomic, proteomic data along with environmental factors.
Integrating data from different sources brings many challenges. In dealing with heterogeneous data, for example, one needs to convert data from different sources into a common format and common dimension. Genomic data arises in the form of vectors, graphs, or sequences, therefore it is essential to carefully consider strategies that best capture the most information contained in each data type before combining them.
Moreover, data from different sources might have different quality and informativity. Probe design and experimental conditions are known to influence signal intensities and sensitivities for many high-throughput technologies [13, 14]. Even for similar data types, data from different sources might have different quality depending on the experimental conditions that generated the data. In microarray experiments, for instance, lack of standards generates heterogeneous datasets for which direct comparison and integration is not possible [15]. Data from different sources might also have different informativity even if their quality is good and reliable; thus one source of data might give us more information than the other in answering the biological question of interest. For example, gene expression microarray data is expected to provide more information in recognizing ribosomal proteins than protein-protein interaction data. However, expression data is not expected to provide much information in identifying membrane proteins [9].
The overarching goals of data integration are to obtain more precision, better accuracy, and greater statistical power than any individual dataset would provide. Moreover, integration can be useful in comparing, validating, and assessing results from different studies and datasets. It is likely that whenever information from multiple independent sources agree, it is more likely for the findings to be valid and reliable than information from a single source [8].
Current methods for data integration in general and combining genomic and genetic data in particular are scattered in the literature and lack solid conceptual framework. Putting them under a single framework would bring more understanding and clarity for the research community. With this background in mind, the objective of this paper is two fold. The first objective is to introduce a conceptual framework for integrating genomic and genetic data. This framework, which can be adapted to most data integration tasks in the life sciences, can serve as a guideline for understanding key issues and challenges that arise in data integration in genetics and genomics. We also believe that the framework we introduce here can be used for motivating and developing improved methods for integrating genomic and genetic data. The second purpose of the paper is to review some of the most commonly used current methods and approaches for combining genomic data. The reviews are done from a statistical perspective and our discussions are focused more on methodological issues and challenges. This could be useful in identifying research directions and might lead to improved methodologies in combining genomic data.
The paper is organized as follows. In Section 2, we provide a conceptual framework for data integration and discuss key concepts regarding this framework. In Section 3, we discuss some of the methods used in integrating similar data types, and methods for integrating heterogeneous data types, including integrating statistical results with biological data, are reviewed in Section 4. A brief discussion and some highlights for future research directions are presented in Section 5.
2. A Conceptual Framework for Data Integration
The concept of data integration is not well defined in the literature and it may mean different things for different people. For instance, Lu et al. defined data integration, in the context of functional genomics, as the process of statistically combining data from different sources to provide a unified view of the whole genome and make large-scale statistical inference [16]. We view data integration in a much broader context so that it includes not only combining of data using statistical approaches, but also data fusion with biological domain knowledge using a variety of bioinformatics and computational tools.
In this section, we propose a conceptual framework for integrating genomic and genetic data. This framework attempts to capture the fundamental aspects of data integration and is developed taking the key steps involved in genomic and genetic data fusion into consideration. A flowchart describing the conceptual framework is given in Figure 1. Below we briefly discuss each of the three key components of data integration: posing the statistical/biological problem; recognizing the data type; stage of integration.
Conceptual framework for data integration in genetics and genomics.
2.1. Posing the Statistical/Biological Problem
Identifying the statistical or biological problem is the first step in any statistical research in general and in genomic and genetic data fusion in particular. Different directions in the framework and methods are followed depending on the biological question of interest. For example, one might merge preprocessed and transformed (independently or in parallel) microarray data from different labs (experiments) to increase sample size and answer a scientific question related to the detection of differentially expressed genes across a range of experimental conditions [17]. Traditional biological research questions are for the most part hypothesis-driven where one performs experiments to answer specific biological hypotheses. However, current high throughput data have a wealth of information in answering many other statistical or biological questions. In modern genomics, it is increasingly accepted to generate data in a relatively hypothesis-free setting where different questions can be posed on the pool of data and data are mined with a variety of computational and statistical tools with the hope of discovering new knowledge.
2.2. Data Types
Current data integration methods fall into two different categories—integrating similar data types (across studies) or integrating heterogeneous data types (across studies as well as within studies). Once we identify the biological or statistical question, we can ask ourselves what type of data we have. Classifying data as similar or heterogeneous is not an easy task. In this paper, we consider data as of “similar type” if they are from the same underlying source, that is, if they are all gene expression, SNP, protein, copy number, sequence, clinical, and so on. We refer to data as of the “heterogeneous type” if two or more fundamentally different data sources are involved. One might, for example, want to develop a predictive model based on different genomic data (SNP, gene expression, protein, sequence) as well as clinical data. These data sets might have different structures, dimensions, and formats. Some of them are sequences, some graphs, and yet others may be numerical quantities. Integration of heterogeneous data, therefore, entails converting each of the separate sources into common structure, format, and dimension before combining them.
Whether data are of similar or heterogeneous type, the issue of quality and informativity is of great importance as well. Each data source is subject to different noise levels depending on the technology, the platform, the lab, and many other systematic and random errors. Therefore, the concept of weighting the data sources with quality and/or informativity scores becomes an essential component of the framework.
2.3. Stages of Integration
Data from different sources can be integrated at three different stages—early, intermediate, or late. The stage at which data are combined depends on the biological question, the nature, and type of data as well as the availability of original data. Regardless of the biological question at hand (e.g., test for differential expression, class discovery, class prediction, gene mapping, etc.) one might, for example, merge data from different studies, experiments, or labs to increase sample size. This is considered as integration at early stage. Merging weighted (by quality and/or informativity scores) data is also considered as early integration. This is because attaching of weights to the data does not change the general format and nature of the resulting data. However, the integration is considered as intermediate if we transform individual data sources into another format before we combine them. For example, in class prediction problems, one might convert the data into similarity matrices such as the covariance or correlation matrix and combine these similarity matrices for better prediction. Unlike the early stage integration, original data sets from the different sources are converted to a common format and dimension. Integration is considered to be at a late stage if final statistical results from different studies are combined. This stage includes, among others, meta-analytic techniques where one typically combines effect sizes or p values across studies.
2.4. Preprocessing
Genomic data are subject to different noises and errors, and a number of critical steps are required to preprocess raw measurements. An important step considered in our framework, therefore, is preprocessing. However, this is not the main focus of this paper and hence we do not go into details. We refer the reader to [18, 19] for more details.
Preprocessing precedes data integration and may include background correction, normalization, and quality assessment of data from high throughput technologies [19]. Approaches for preprocessing vary depending on the type and nature of data. Preprocessing methods for microarray data are, for example, different from that for array CGH or proteomic data. Moreover, data from different technologies and platforms might be preprocessed differently. For example, different approaches are utilized for preprocessing cDNA and Affymetrix gene expression microarray data [18, 19].
Data preprocessing can be done at any step of the data integration process. Some form of preprocessing is almost always done at the initial stage [13]. However, genomic data, in most cases, has to go through some sort of preprocessing before doing any statistical analysis to answer the biological question of interest. There is a large body of literature on this topic, and several ways of preprocessing high throughput data have been proposed [18, 19]. Approaches, both graphical and statistical, are also available for visualizing and checking if data needs to be preprocessed. In microarray studies, the standard procedure for researchers is to use preprocessed data as the starting point; however, this has prevented many researchers from carefully considering their preprocessing methods [20].
Data matching is another preprocessing step that needs to be taken into account before combining data from high throughput technologies. In gene or marker specific data integration, a major challenge in pooling information from different data sources can be partly due to the fact that measurements are obtained by different technologies or they measure different aspects of the same underlying quantity. For example, Affymetrix uses different numbers of probes to measure the same gene in different chip types. Therefore, it is impossible to get comparable gene expression levels across different chip types simply based on gene identifiers since they used different probe sequences for the given target probes. Mecham et al. [21] proposed a sequence-based matching of probes instead of gene identifier-based matching. The results showed that at different levels of the analysis (e.g., gene expression ratios), cross-platform consistency is significantly improved by sequence-based matching.
Combining data from different genotyping projects presents a problem in that different marker sets are used for different arrays. For example, out of the approximately 1 million SNPs on each of the Affymetrix Human SNP 6.0 and Illumina Human1M arrays, only about 250,000 SNPs are common to both assays [22]. To overcome this difficulty, genotype imputation algorithms, such as MACH [23], IMPUTE [24], and fastPHASE [25], have been developed to impute alleles at ungenotyped markers, based on the genotypes of surrounding markers. In summary, the preprocessing step can hugely affect the properties of the final statistical summaries and hence the statistical results. Therefore, methods for preprocessing must be chosen with care.
3. Integrating Similar Data Types
Although posing the statistical/biological problem is the first step in any study involving data integration, data integration in general is divided into two broad categories: integration of similar data types and integration of heterogeneous data types. In this section, we give a review of some of the current methods available for integrating similar data types. Similar data types have been combined to answer different biological questions, and approaches for integrating such data at early, intermediate, or late stages have been proposed in literature. However, these approaches are in general meta-analytic methods. Some examples from the published literature that can serve as illustration for data integration concepts corresponding to different biological/statistical questions, data types, and stages of integration are highlighted in Table 1.
Some illustrative examples for integrating similar and heterogeneous genomic, genetic, and proteomic data.
3.1. Integration of Linkage Studies
Linkage analysis is a gene mapping technique which is based on the process of recombination, or the crossing over of parental
chromosomes when forming gametes which will eventually be passed on to offspring. In order to observe or infer recombination
events, families are required to reconstruct the transmission of alleles and phenotypes through several generations. Highly
polymorphic markers are genotyped either in a region of interest, based on previous knowledge or hypotheses (candidate gene
mapping), or across the entire genome (genome-wide scans). Linkage analysis looks for the cosegregation of a marker and the
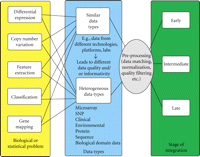
trait of interest in each family, and one way to assess linkage statistically is through the log odds, or LOD score:
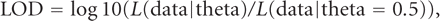 where L is a likelihood function and theta is a measure of the amount of recombination between the marker and the trait. The LOD
score can be maximized over a grid of theta values, but is usually just computed at fixed theta intervals. Scores for each
family are summed, and an overall LOD score > 3 (i.e., likelihood ratio than 1000:1 odds) is generally considered to be a
significant evidence for linkage. For further details, the interested reader is referred to an excellent textbook in [30].
where L is a likelihood function and theta is a measure of the amount of recombination between the marker and the trait. The LOD
score can be maximized over a grid of theta values, but is usually just computed at fixed theta intervals. Scores for each
family are summed, and an overall LOD score > 3 (i.e., likelihood ratio than 1000:1 odds) is generally considered to be a
significant evidence for linkage. For further details, the interested reader is referred to an excellent textbook in [30].
Linkage analysis has been very successful for rare Mendelian disorders, that is, diseases where the variant is associated with a large increase in risk, and usually only one variant is responsible for the phenotype. However, in the case of complex traits, where multiple variants are likely to contribute, each with more modest risk, large collections of families need to be genotyped and phenotyped to have modest power to detect linkage, which is a costly and time-consuming undertaking. Combining information from several scans can, therefore, help overcome these difficulties. Three main strategies have been used to integrate linkage data: pooling of datasets (integration at an early stage), combining linkage statistics or P-values (integration at a late stage), and combining effect sizes (also integration at a late stage).
When the raw data from all studies are available, the most powerful approach is to simply pool the datasets and analyze this large dataset as if it were one study, termed a “mega analysis” [31]. This assumes that the same markers are common to all studies, and that there were identical ascertainment strategies and similar allele frequencies in the populations from which the samples were derived from. It is also equivalent to simply sum the LOD scores from various studies, under the same restrictions [32]. Since it is rare to have identical marker maps across studies, methods to allow LOD score calculations at arbitrary marker positions were developed to overcome this problem [33, 34]. An alternative method, known as the posterior probability of linkage (PPL), puts the question of linkage in a Bayesian context, which allows the posterior distribution of linkage, given the data, to be updated as new data and studies are accumulated [35].
In the more common situation when the raw data is not available, perhaps the simplest method to combine independent P-values is the one developed by Fisher [36],
 where k is the number of studies,
where k is the number of studies, 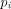 is the P-value obtained from study i. The statistic S is asymptotically distributed as a chi-square with
is the P-value obtained from study i. The statistic S is asymptotically distributed as a chi-square with 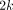 degrees of freedom. This procedure was used by Allison and Heo [37] to identify regions linked to obesity, and a modified version was used by Badner and Gershon [38] for autism studies. The null hypothesis for this test is that none of the studies show significant results, against the
alternative that at least one is significant. This may not be the question that a researcher is interested in asking; a more
relevant question may be whether all studies support a common hypothesis. In this situation, Rice (1990) [39] suggests that a summary statistic based on the mean of the normal-transformed P-values may be more appropriate.
degrees of freedom. This procedure was used by Allison and Heo [37] to identify regions linked to obesity, and a modified version was used by Badner and Gershon [38] for autism studies. The null hypothesis for this test is that none of the studies show significant results, against the
alternative that at least one is significant. This may not be the question that a researcher is interested in asking; a more
relevant question may be whether all studies support a common hypothesis. In this situation, Rice (1990) [39] suggests that a summary statistic based on the mean of the normal-transformed P-values may be more appropriate.
In the Fisher method, a single P-value per study is used. However, in the context of a genome-wide scan, many markers are genotyped, and so many LOD scores or P-values are calculated. To address this issue, Wise et al. [40] proposed a method called genome search meta-analysis (GSMA) which ranks significant results within each study, and sums the ranks for each region across all studies. The ranks can be weighted by study characteristics such as the number of pedigrees or the number of markers. This test will detect regions which are implicated in several studies.
The use of P-values from individual studies generally precludes the estimation of average effect sizes, which can be of interest in linkage studies. These estimates are the main goal of most standard meta-analyses. Li and Rao [41] proposed the use of a random effects model [42] to combine regression coefficients from a linear model of the squared trait differences as the dependent variable, and the proportion of alleles shared identical by descent (IBD) at a marker for sibling pairs as the independent variable. The model was also applied using the proportion of alleles shared IBD directly as the measure of effect size [43]. Along with an overall estimate of effect size, the random effects model has the advantage of being able to test for and control for differences in effect sizes across studies (sometimes referred to as heterogeneity).
3.2. Integration of Genetic Association Studies
In the past few years, it has been shown that genome-wide association studies have strong power to identify genetic determinants for common and rare diseases. Due to the high cost of performing these types of studies, it becomes more and more important to integrate evidence from multiple studies in characterizing the genetic risks of these diseases. Meta-analyses can offer both enhanced power to detect associations and increased precision of estimates of its magnitude. There are two major methods with focusing on late stage integration. One is combining effect sizes, primarily the odds ratio (OR) and another is to combine P-values [26, 44, 45]. The effect size based method can be fixed effects or random effects models. For example, Ioannidis et al. [26] applied a random effects model to combine all data sets generated in three stages from three genome-wide association (GWA) studies on type 2 diabetes. Details of the design and populations of these studies have been presented in the original publications [46–48]. Ioannidis et al. [26] selected 11 polymorphisms suggested as susceptibility loci for type 2 diabetes in at least one of the three studies. They found 5 of the 11 genetic variants have moderate to very large heterogeneity across studies. Therefore, they used random effects calculations incorporating between study heterogeneity for these 5 polymorphisms and found more conservative P-values for the summary effects compared with the fixed effects calculations. Instead of focusing on meta-analysis of only identified polymorphisms, Zeggini et al. [45] applied fixed effects model and combing P-value methods to meta-analyze the same data sets, they detected at least six previously unknown loci with robust evidence for association.
3.3. Integration of Gene Expression Microarray Studies
Microarrays have been widely used in identifying differentially expressed genes [48, 49] and for building gene expression profile-based predictors for disease outcome diagnosis [50–54]. Although some of these studies have led to promising results [51], it is difficult to directly compare the results obtained by different groups addressing the same biological problem. This is because laboratory protocols, microarray platforms, and analysis techniques used in each study may not be identical [55, 56]. Moreover, most individual studies have relatively small sample sizes, and hence predictive models trained by individual studies using cross-validation are prone to over-fitting, leading to prediction accuracies that may be less robust and lack generalizability [57]. Recent studies show that systematic integration of gene expression data from different sources can increase statistical power in detecting differentially expressed genes while allowing for an assessment of heterogeneity, and may lead to more robust, reproducible, and accurate predictions [12, 15, 17, 56, 58–62]. Therefore, our ability to develop powerful statistical methods for efficiently integrating related genomic experiments is critical to the success of the massive investment made on genomic studies. Here, we highlight some of the strategies that have been used to integrate microarry gene expression studies.
Combining gene expression data at early and late stages has been considered by different groups. In integrating gene expression data at an early stage, data sets generated in each study are first preprocessed independently or in parallel, and then the preprocessed datasets are put together so that the integrated data set can be treated as one data set. In this way, the sample size of the study is greatly increased. Several transformation methods have been proposed to process gene expression measures from different studies [17, 56, 59, 62]. For example, Jiang et al. [17] transformed the normalized data sets to have similar distributions and then merged the transformed data sets. Wang et al. [59] standardized each gene expression level based on the average expression measurements and the standard errors estimated from prostate cancer samples. These methods are simple and in many cases, if the transformation is carefully made, lead to improved prediction [17]. Nevertheless, there are no consensus or clear guidelines as to the best way to perform such data transformations.
At the late stage, results from statistical analyses are combined using meta-analytic approaches. Similar to the case of linkage and association gene mapping studies, one of the popular approaches combines effect sizes from different studies while taking interstudy variability into account when estimating the overall mean for each gene across studies. For example, Choi et al. [15] focused on integrating effect size estimates in individual studies into an overall estimate of the average effect size. The effect size was used to measure the magnitude of treatment effect in a given study and random effects model was adopted to incorporate interstudy variability. Using the same microarray data sets as those used by Rhodes et al. [12], Choi et al. [15] demonstrated that their method can lead to the discovery of small but consistent expression changes with increased sensitivity and reliability among the datasets. For each gene, the widely used effect size measure is the standardized mean difference which is obtained by dividing the difference in average gene expression between groups of interest by a pooled estimate of standard deviation [63, 64]. It is well known in microarray data analysis that the estimated standard deviation might be unstable when the sample size in each group is small. Therefore, much effort has been made to overcome the shortfall by using a penalty parameter for smoothing the estimates using information from all genes rather than relying solely on the estimates from an individual gene [4, 65].
As mentioned before, the other meta-analytic technique commonly used combines P-values across different studies. For example, Rhodes et al. [12, 66] integrated results from prostate cancer microarray studies which have been performed on different platforms. Differential expression was first assessed independently for each gene in each dataset using P-values and P-values from individual studies were combined using Fisher's method (see also Section 3.1). Their analysis revealed stronger evidence for statistical significance from the combined analysis than any of the individual studies separately. Combining P-values can be useful in detecting effects with improved statistical significance, but this method does not indicate the direction of significance (e.g., up- or downregulation) [67]. Instead of integrating P-values directly, some studies explored combining the ranks of the P-values from different studies [61, 68]. For example, DeConde et al. [61] proposed a rank-aggregation method and combined microarray results from five prostate cancer studies where they showed that their approach can identify more robust differentially expressed genes across studies.
The data integration approaches discussed above to integrate microarrays are in a quality-unweighted framework [12, 15, 17, 56, 58, 59, 61, 62, 66]. However, it has been argued that studies of higher quality give more accurate estimates and, as a result, should receive higher weight in the analysis summarizing findings across studies [69]. In gene expression microarrays, many genes may be “off” or not detectable in a particular adult tissue, moreover, some genes may be poorly measured due to probes that are not sufficiently sensitive or specific. Therefore, the signal strength and clarity will vary across the genes, suggesting that a quality measurement could highlight strong and clear signals [70, 71]. How to best measure the quality of a gene expression measurement and how best to use such a quality measure are still open research questions. However, different strategies can be considered for incorporating quality weights into meta-analysis of microarray studies. For example, a quality threshold can be defined and only genes that are above this threshold can be included in the meta-analysis. However, the choice of threshold will be arbitrary. In a recent study, our group proposed a quality measure based on the detection P-values estimated from Affymetrix microarray raw data [60, 70]. Using an effect-size model, we demonstrated that the incorporation of quality weights into the study-specific test statistics, within a meta-analysis of two Affymetrix microarray studies, produced more biologically meaningful results than the unweighted analysis.
4. Integrating Heterogeneous Data Types
Perhaps the most challenging type of data integration is combining heterogeneous data types. A wide variety of genomic and proteomic data are becoming available at an unprecedented pace including data on, but not limited to, gene expression (quantitative numbers), gene/protein sequences (strings), gene-gene/protein-protein interactions (graphs). There is also a growing interest for integration of these and related molecular information with clinical, laboratory, as well as environmental data. Broadly speaking, integrating heterogeneous data types involves two steps. The first one is converting data from different sources into a common format. The second, equally important, step is to combine the data and perform statistical analysis on the combined data set. Here we survey some of the currently available approaches for integrating heterogeneous data types. Some illustrative examples are highlighted in Table 1. An illustrative flowchart outlining integrative analyses of heterogeneous data for finding disease-causing genes is shown in Figure 2.
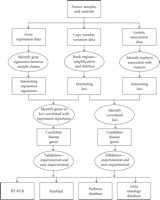
An illustrative flowchart for finding disease causing genes by integrating heterogeneous data.
4.1. Integration of Gene Expression with Genotype Data
Gene expression levels of many genes have been successfully used to show natural variation in humans [72, 73]. Using regression analysis where the dependent variables are expression levels and the independent variables are the genotypes, it has been shown that expression levels may be influenced by single nucleotide polymorphisms [72–75]. These mapping efforts have identified quantitative trait loci (QTLs) that may be in the gene's own regulatory regions (cis-acting QTLs) as well as elsewhere in the genome (trans-acting QTLs) using both linkage [72] and association analysis [73, 74]. For the association analysis, Stranger et al. [74] examined all possible combinations of gene expression phenotype/marker genotype combinations, whereas Cheung et al. [73] examined only gene expression phenotype/genotype combinations under linkage peaks identified in the study by Morley et al. [72]. However, it may be possible that multiple loci play a role in regulating the expression level of a single phenotype. To this effect, our group used the gene expression and SNP data reported in Morley et al. [72] and applied stepwise regression analysis to look for additive effects of the SNPs which led to the identification of cis-and trans-acting loci that regulate gene expression [75]. We identified many expression phenotypes that have significant evidence of association and linkage to one or more chromosomal regions and showed that much of the observable heritability in the phenotypes could be explained by simple SNP associations.
Due to the large number of genes in current high volume data sets and the existence of various degrees of noise in the data, integration involving all single-nucleotide polymorphism (SNP) loci and gene expression phenotypes may computationally challenging, and results may lack biological plausibility and interpretability. One promising approach, that is computationally efficient and can lead to more robust and interpretable results, is to use methods that induce sparseness in the integrated solutions where noisy data are automatically filtered out from analysis. For example, our group has recently introduced a novel sparse canonical correlation analysis (SCCA) statistical method, which allowed us to examine the relationships of many genetic loci and gene expression phenotypes by providing sparse linear combinations that include only a small subset of loci and gene expression phenotypes [76]. The correlated sets of variables resulting from sparse canonical correlation analysis are sufficiently small for biological interpretability and further follow up. We applied SCCA to data reported in [72] and identified small but interesting group of loci and gene expressions that have maximal correlation across the two data sources (gene expression and genotypes).
4.2. Integration of Copy Number Variation and Gene Expression Data
Array CGH (aCGH) microarray technology has been widely used to identify small or large regions of amplifications and deletions along the genome of organisms. Recent studies have tried to incorporate gene expression data with aCGH data for finding disease causing genes [27, 77–79]. For example, Pollack et al. [77] analyzed gene expression levels in parallel with copy number aberrations for the same set of breast tumors. They found that DNA copy number does have an impact on gene expression levels, and that a 2-fold change in DNA copy number corresponds to an average 1.5-fold change in expression level [72–75]. However, it has also been observed that many overexpressed genes were not amplified and that not all amplified genes were highly expressed, but the small number of genes that were overexpressed and amplified could be interesting genes. For example, Platzer et al. [80] measured gene expression levels and DNA copy numbers of colon cancer samples, and found four chromosomal arms that contained amplifications in most samples. Among expression levels of 2146 transcripts on these arms, only 81 have greater than 2-fold change in gene expression. They concluded that chromosomal amplifications do not result in global over expression of the genes located at that position. Huang et al. [79] also found that genomic DNA copy number aberrations (amplification or deletion) appeared not to be parallel with the corresponding gene expressions in any given samples. Most of these methods explore the relationship of genomic DNA copy number and gene expression at relatively the same positions of the genes on the genome. However, since it is known that genes on a chromosome are coregulated [81], a better way is to determine clusters of significantly over or under expressed genes by taking the chromosome position into account. This can also be applied on CGH data, and then a correlation of the clustered or coregulated expression signatures and copy number data can be determined [78].
4.3. Kernel Based Data Integration for Class Prediction
One of the novel and most promising methods for integrating heterogeneous data types are kernel-based statistical methods. In kernel-based methods, each data set is represented by a so-called kernel matrix, which essentially constitutes similarity measures between pairs of entities (genes, proteins, patients, etc.). In general, these methods are applied in biological problems related to class discovery and class prediction. In functional genomics, for example, one is interested in discovering new functional classes and/or assigning each gene or protein into already existing classes. It is also useful in cancer research where one is interested in discovering new tumor subtypes and/or assigning patients into already existing tumor types. Kernel-based statistical methods are tools which have already proven to be powerful in areas such as pattern recognition, chemoinformatics, computational linguistics, and bioinformatics. Their rapid uptake in these applications is due to their reliability, accuracy, and computational efficiency as well as their ability to handle various types of data [82].
One can describe kernel-based statistical learning approaches using two basic steps. The first one is choosing the right kernel for each data set. This is a crucial and difficult step in combining heterogeneous data using kernels. One of the reasons for the success of kernel methods is that kernel matrices can be defined for any type of data as well as their ability to incorporate prior knowledge through the kernel function [82]. The choice of kernel matrices depends on what type of data we have (e.g., diffusion kernel for graphical data and sequence-based kernels based on algorithms such as BLAST for protein sequences) and if the patterns in the data are linear (linear or standardized linear kernel can be used) or nonlinear (Gaussian or other nonlinear kernels can be used). One can also define different kernels on the same data sets. This allows us to get different view of the same data set and might provide us more information than using a single kernel. For example, Lanckriet et al. [9] defined 7 different kernels on three different data sources for predicting membrane and ribosomal proteins. The second equally important and challenging step deals with combining the kernels from the different data sources to give a complete representation of available data for a given statistical task. Basic mathematical operations such as multiplication, addition, and exponentiation preserve properties of kernel matrices and hence produce valid kernels. The simplest approach is to use the sum of the different kernels, which is equivalent to taking the average kernel. This naïve combination has been used mainly for comparison purposes. However, not all data have equal quality and informativity. Depending on the statistical and biological question at hand, data from one source might contain more information than the other. Moreover, the quality of data might vary because of different limitations and factors involved in different experiments. To our knowledge, currently there are no published methods that explicitly incorporate quality and informativity measures into the kernel framework.
There are few kernel-based statistical methods proposed for integrating heterogeneous genomic data. Lanckriet et al. [9] used kernel-based support vector machine (SVM) method to recognize particular classes of proteins—membrane proteins and ribosomal proteins. Their method finds the classification rule as well as the corresponding weights for each data set. The performance of the SVM trained on the combined data set is better than that of the SVM trained on each of the individual data sets. Moreover, the weights produced from the algorithm give some measure of the relative importance of the different data sets. Another similar kernel-based approach was used by Daemen et al. [4, 10]. They used kernel-based least square support vector machine (LS-SVM) to combine clinical and microarray data [10]. The same group applied their method to combine microarray and proteomics data [4]. They chose a standardized linear kernel for both data sets in both papers. In the first paper, leave-one-out cross validation was performed on the training data set to get optimal weights. The model based on the clinical and microarray data performed slightly better than the model based on each of the data sets alone. The performance of their method was also compared with three conventional clinical prognostic indices and was shown that the kernel-based integrated microarray and clinical data outperforms all three conventional approaches. In the second study, the authors used the same method to combine microarray and proteomic data to predict the response on cetuximab in patients with rectal cancer. Tissue and plasma samples were taken from the patients before treatment and at the moment of surgery. Tissues were used for microarray analysis and plasma samples were used for proteomics analysis. They defined four kernels from these data sets and assigned equal weights to each one of them, that is, a naïve combination of kernels was used. The method trained on microarray data (with 5 genes) and protein data (10 proteins) performed better than any of the other alternatives they considered.
4.4. Integrating Statistical Results with Biological Domain Data
The ultimate purpose of statistical analysis on genomic data is to gain some insight into the fundamental biology. Annotation of statistical results helps biologists in interpreting discovered patterns. A wide variety of biological information is available to the public, such as information on published literature on the topic of interest (e.g., PubMed) and functional/pathway information (e.g., Gene Ontology, KEGG). Integrating biological information with statistical results is, therefore, another important type of data integration which can be considered as a bridge between statistical results and biological interpretation. Including biological domain data in statistical analysis can be done at any stage of analysis. Al-Shahrour et al. [29], for example, combined statistical results from gene expression data with biological information in discovering molecular functions related to certain phenotypes. Another popular approach to incorporate prior biological knowledge into statistical analysis is gene set enrichment analysis (GSEA) [83, 84]. Given an a priori defined set of genes, the goal of GSEA is to determine whether a particular gene is enriched or not, that is, whether members are randomly distributed throughout or primarily found at the top or bottom of a ranked list of gene differential expression results.
There is also a rapidly growing list of computational and visualization tools that can be used to integrate statistical findings with biological domain information, and thereby facilitating interpretation. For instance, packages from the bioconductor project (http://www.bioconductor.org) provide powerful analytical annotation and visualization tools for a wide range of genetic and genomic data sets.
5. Summary and Future Directions
With a rapidly increasing amount of genomic, proteomic, and other high throughput data, the importance of data integration has increased significantly. Biologists, medical scientists, and clinicians are also interested to integrate recently available high throughput data with already existing clinical, laboratory, as well as prior biological information. Moreover, data have been produced in various formats (graphs, sequences, vectors, etc.) and dimensions and, as a result, a simple merge of available data is not applicable and in some cases impossible. Furthermore, data from different sources are subject to different noise levels due to difference in technologies, platforms and other systematic or random factors affecting the experiments. Consequently, data might have different qualities and a naïve combination of data is not appropriate in such cases. The concept of data informativity is also essential in any data integration problem. Data from various sources might contain different informativity for a given statistical or biological task. One data source might, for example, be more informative than the other. A good data integration method should, therefore, take these into account. Even if quality scoring has been used in traditional statistical analysis, use of quality weights is not common in genetics and genomics. Moreover, appropriate quality and informativity measures have not been defined for many data types. An extensive research is, therefore, needed in developing quality and informativity scores for various genomic, genetic, and proteomic data.
In this paper, we proposed a conceptual framework for genomic and genetic data integration. This framework, with a little modification, can also be useful in any data integration problem. The framework provides different steps involved in genomic data integration and addresses different issues and challenges. Moreover, putting current methodologies for data integration under a single framework brings more understanding in the research community. Furthermore, we hope that it would play an important role in the development of standardized and improved data integration methods that takes the quality, informativity, and other aspects of individual data sets.
Acknowledgments
This work was partially supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Mathematics of Information Technology and Complex Systems (MITACS), the Canadian Institute of Health Research (CIHR) (grant number 84392), and Genome Canada through the Ontario Genomics Institute. The auhtors would also like to thank two anonymous reviewers for helpful comments.
- Received September 25, 2008.
- Accepted December 1, 2008.
- © 2009 Jemila S. Hamid et al.















