|
|
PAGES DU PRATICIEN
Susceptibilité génétique au cancerLe rôle des polymorphismes dans les gènes candidats
Linda M. Dong, MPH, PhD;
John D. Potter, MD, PhD;
Emily White, PhD;
Cornelia M. Ulrich, PhD;
Lon R. Cardon, PhD;
Ulrike Peters, PhD, MPH
JAMA. 2008;299(20):2423-2436
RÉSUMÉ
| |
Contexte Les progrès continus des technologies de génotypage et l'inclusion du recueil de l’ADN dans des études d'observation ont permis un nombre croissant d'études génétiques d'association.
Objectif Evaluer le progrès global et la contribution des études d'association des gènes candidats à la compréhension actuelle de la susceptibilité génétique au cancer.
Sources des données Nous avons systématiquement examiné les résultats des méta-analyses et d’analyses groupées sur les polymorphismes génétiques et le risque de cancer publiées jusqu'en mars 2008.
Sélection des études Nous avons identifié 161 méta-analyses et analyses groupées, incluant 18 localisations de cancer et 99 gènes. Les analyses devaient répondre aux critères suivants : inclusion d’au moins 500 cas, risque de cancer comme critère, ne pas s’être concentrée sur les marqueurs génétiques d'antigènes HLA, et avoir été publiées en anglais.
Recueil des données Les informations sur le site du cancer, le nom du gène, la variante, l'évaluation du paramètre et l'intervalle de confiance à 95% (IC), la fréquence allélomorphe, le nombre d'études et de cas, les essais d'hétérogénéité de l’étude, et le biais de publication ont été extraits par un investigateur et passés en revue par les autres investigateurs.
Résultats Ces 161 analyses ont évalué 344 associations de cancer et de variante génétique et inclus en moyenne 7.3 études et 3551 cas (extrêmes, 508-19729 cas) par association étudiée. Les rapports récapitulatifs de cotes (OR) pour 98 (28%) associations statistiquement significatives (P value<0.05) ont été ensuite évalués en estimant la probabilité d’un rapport faux-positif (FPRP) à une probabilité antérieure donnée et à une puissance statistique donnée. À un niveau de probabilité antérieure de 0.001 et à la puissance statistique permettant de détecter un OR de 1.5, 13 associations cancer-variant génétique sont demeurées notables (FPRP<0.2). En assumant une probabilité antérieure très basse de 0.000001, semblable à une probabilité présumée d’un polymorphisme simple d’un nucléotide aléatoirement choisi dans une étude d'association génomique large, avec une puissance statistique pour détecter un OR de 1.5, 4 associations ont été considérées notables en ayant une valeur <0.2 de FPRP : GSTM1 nul et cancer de la vessie (OR, 1.5 ; IC 95%, 1.3-1.6 ; P=1.9x10–14), acétyleur lent NAT2 et cancer de la vessie (OR, 1.46 ; IC 95%, 1.26 - 1.68 ; P=2.5 x 10–7), MTHFR C677T et cancer gastrique (OR, 1.52 ; IC 95%, 1.31 - 1.77 ; P=4.9x10–8), et GSTM1 nul et leucémie aiguë (OR, 1.20 ; IC 95%, 1.14 - 1.25 ; P =8.6x10–15). Lorsque l’OR utilisé pour déterminer la puissance statistique a été abaissé à 1.2, 2 des 4 associations notables ont persisté: GSTM1 nul avec le cancer de la vessie et la leucémie aiguë.
Conclusion Dans cette revue d’études d'association de gène candidat, presque un tiers des associations cancer-variant du gène était statistiquement significatif, les variants des gènes codant le métabolisme des enzymes étant parmi les associations les plus consistantes et les plus fortement significatives.
Au cours des dernières décennies, un vaste effort a été fait pour identifier des sources de susceptibilité génétique au cancer. L’International Human Genome Sequencing Project et le International HapMap Project ont produit un très grand nombre de données sur la localisation, la quantité, le type, et la fréquence des variants génétiques dans le génome humain.1-4 Facilité par les progrès technologiques en cours qui permettent résultats de génotypage plus rapide et meilleur marché, un nombre important et croissant d'études d'observation évaluant l'association entre les variants des gènes candidat et le risque de cancer a émergé.5
Ce nombre croissant d'études nous a incités à évaluer la contribution globale de ces études à notre compréhension actuelle de la susceptibilité génétique au cancer. Une des critiques principales de l'épidémiologie génétique a été un manque de reproductibilité. Il existe plusieurs exemples d’études explorant une conclusion statistiquement significative précédemment publiées d'un variant génétique avec cependant une absence de reproductibilité de ces résultats, ce qui suggère un grand nombre de rapports faussement positifs. 6,7 La taille de ces études génétiques d'association est également une question méthodologique importante qui a incité à l'utilisation de méta-analyses et d’analyses groupées pour combiner des résultats statistiquement significatifs et non significatifs d’études individuelles avec une pondération de ces résultats par leur précision (une fonction de la dimension de l'échantillon).8-10
Pour évaluer le progrès global des études d'association des gènes candidats en identifiant les variants génétiques associés au risque de cancer, nous avons systématiquement examiné les résultats de toutes les méta-analyses publiés et des analyses groupées sur les polymorphismes génétiques et le risque de cancer et rapportons les points d’estimation observés, les intervalles de confiance à 95% (IC) et les valeurs de P. Comme 3 paramètres sont juste nécessaires pour évaluer entièrement les tests médicaux diagnostiques (spécificité, sensibilité, et valeur prédictive d'un test positif), 3 paramètres analogues sont nécessaires pour évaluer entièrement les tests statistiques d'une association (par exemple, entre un variant génétique et un cancer).11 La valeur de P, la probabilité d'obtenir une estimation plus extrême que celle observé quand l'hypothèse nulle d’absence d’association (rapport de cotes [OR], 1.0) est réelle, est analogue à 1 moins la spécificité (la probabilité d'un essai classant une personne comme ayant la pathologie quand elle n'a vraiment pas la pathologie). La puissance de l’étude, la probabilité de détecter une association quand elle existe, est analogue à la sensibilité (la probabilité d'un essai classant quelqu'un comme ayant une affection quand elle l'a vraiment.) Cependant, il est bien établi dans les diagnostics médicaux que la spécificité et la sensibilité peuvent être élevées, mais la valeur prédictive d'un test positif peut encore être basse. Ceci parce que, si la condition est rare, les résultats positifs des examens diagnostiques seront la plupart du temps des faux positifs. Ceci est moins apprécié mais également important lors de l’évaluation des tests statistiques des associations présumées : lorsque la probabilité antérieure est faible qu'une hypothèse d'exposition à une maladie soit vraie, une conclusion statistiquement significative a alors une possibilité élevée d'être un faux positif. La probabilité d’un rapport faux positif (FPRP) est définie comme « la probabilité d’absence d’association compte tenu d’une conclusion statistiquement significative»12 et est analogue à 1 moins la valeur prédictive d'un test positif. Aussi, c'est la FPRP plutôt que la valeur de P qui répond à la question de savoir quelle est réellement la probabilité de l'hypothèse, telle qu’elle est évaluée.
Dans cet article, nous évaluons les résultats des études d'association gène candidat-cancer en présentant la valeur de P, la puissance, et la FPRP pour toutes les associations statistiquement significatives rapportées dans les méta-analyses ou les analyses groupées. La FPRP est calculée à partir de la puissance statistique du test, de la valeur observée de P, et d'une probabilité antérieure donnée pour l'association.12 Puisque les probabilités antérieures ne sont pas facilement déterminées, nous avons calculé la FPRP pour 2 niveaux de probabilité antérieure appropriées pour une gamme d’hypothèses ; de basses probabilités, appropriées pour des polymorphismes avec des conséquences fonctionnelles connues dans des gènes candidats importants, à des probabilités très basses, appropriées pour des variants aléatoirement choisis et utilisées dans les études d'association à l’échelle du génome.
Cette revue présente les informations sur la connaissance produite jusqu'ici par les études d'association de gène candidat entreprises pour identifier des gènes de susceptibilité au cancer et peut également être employée pour diriger les futures études vers des secteurs qui demeurent peu clairs. En outre, les résultats de cette analyse fournissent des informations sur la fréquence allélomorphe et la taille prévue de l’effet (à proprement parler, la force d'association), qui peuvent être utiles pour planifier des études (échelle du génome) d'association.
METHODES
Nous avons identifié les méta-analyses publiées et les analyses groupées ayant évalué l'association entre les polymorphismes génétiques et le risque de cancer dans les études d'observation (cas-témoin et les études nichées cas-témoin) et répertoriées dans PubMed jusqu'au 15 mars 2008. Les méta-analyses et les analyses groupées sont définies par des outils qui intègrent les résultats de différentes études qui, seules, peuvent ne pas avoir de puissance suffisante pour détecter une association statistiquement significative.8-10 En bref, les OR des données (OR bruts et ajustés) utilisés pour une méta-analyse sont extraits de résultats publiés, tandis que les séries de données originaux provenant d'un certain nombre d'études indépendantes sont employées pour les analyses groupées. Nous avons effectué une recherche de la littérature dans la base de données PubMed utilisant les mots clés de recherche suivants pour nos recherches dans la littérature : les combinaisons de mots-clés cancer plus meta plus gene, cancer plus pooled plus gene, cancer plus consortium plus gene, et les combinaisons de mot-clé gene plus cancer et genetic plus cancer limités au type de publication « méta-analyse. »
Nous avons examiné 794 articles identifiés par nos méthodes de recherche, examiné en détail 224 articles, pour finalement inclure 161 articles (FIGURE 1). Les études incluses dans notre revue devaient répondre à tous les critères suivants : (1) inclure au moins 500 cas combinés de toutes les études récapitulées, (2) avoir un risque de cancer comme critère d’évaluation (les analyses de la survie, les marqueurs néoplasiques, ou les précurseurs, tels que des polypes, ont été exclus), (3) exclure les marqueurs génétiques tels que l’antigène HLA, et (4) être publiés en anglais. En outre, parce que cette revue se concentre sur des variants communs, les méta-analyses et les analyses groupées de gènes ayant une basse fréquence, et une haute pénétrance, tels qu’APC, BRCA1 ou BRCA2, ont été exclus. En outre, bien que des associations statistiquement significatives aient été rapportées pour les polymorphismes HRAS1 et le risque de cancer de sein et du poumon, ces associations ont été remises en cause en raison des méthodes de génotypage défectueuses. Aussi, ceux-ci ne sont pas rapportés avec les autres associations statistiquement significatives. Pour éviter la duplication des résultats de plus d’une méta-analyse ou d'une analyse groupée sur la même association, nous avons choisi la publication la plus récente, qui typiquement avait le plus grand nombre de cas (parfois plus faible, en raison de critères plus stricts d'inclusion). Les données extraites à partir de chaque méta-analyse ou d’analyse groupées ont inclus la localisation du cancer, le nom du gène, le variant génétique, le point d’estimation (risque relatif [RR] ou OR) et IC 95%, fréquence allélomorphe (si présente), le nombre d'études, le nombre de cas, le test d'hétérogénéité d'étude (par exemple, test Q), et un test de biais de publication (dont le test de Begg, le test d'Egger et funnel plots). Les évaluations des effets aléatoires des méta-analyses sont présentées, sauf si seules les évaluations des effets fixes étaient disponibles.
|
|
|
Figure 1. Sélection des études
|
|
|
Nous avons calculé des estimations récapitulatives pour décrire les rapports publiés et identifiés par notre recherche. Les différences dans le nombre d'études et de cas ont été évaluées par un test t. Les associations étaient considérées statistiquement significatives si la valeur rapportée de P était <0.05 ou si l’IC à 95% excluait 1.0. La valeur de P était déterminée d'abord en calculant un score z sur la base de l’OR rapporté et de l’IC 95%, z score=ln (OR)/[(ln (IC supérieur) – ln (IC inférieur)/(2 x 1.96)], puis en le comparant alors à une distribution normale.
Pour chaque association statistiquement significative rapportée, nous avons estimé la FPRP suivant les méthodes décrites par Wacholder et coll.12 La valeur de FPRP est déterminée par la valeur de P, la probabilité antérieure donnée pour l'association, et la puissance statistique de l'essai. L'attribution d'une probabilité antérieure doit être déterminée avant d'obtenir les résultats d'une étude et doit être indépendante de n'importe quelles données utilisées dans l'analyse. Les probabilités antérieures sont subjectives et sont influencées par les résultats épidémiologiques précédents et la preuve expérimentale sur les fonctions connues d'un variant génétique. Par conséquent, nous avons choisi de calculer des valeurs de FPRP pour 2 niveaux de probabilités antérieures : à un bas niveau antérieurement qui serait semblable à ce qui serait prévu pour un gène de candidat (0.001) et à un très bas niveau antérieur qui serait semblable à ce qui serait prévu pour un polymorphisme simple aléatoire d’un nucléotide (SNP) (0.000001), permettant ainsi aux lecteurs d'évaluer l'association en utilisant leur propre jugement vis-à-vis des preuves à l'appui pour un locus donné. Wacholder et coll.12 suggèrent d'estimer la puissance statistique en se basant sur la capacité de détecter un OR de 1.5 (ou son réciproque, 0.67=1/1.5 pour des OR<1.0), avec un niveau  égal à la valeur de P observée. 12 Mais compte tenu de l'attention récente portée à des OR beaucoup plus bas cette évaluation peut être trop conservatrice ; aussi, nous avons choisi de présenter les résultats pour des OR de 1.5 et 1.2 (ou leur réciproque 0.83=1/1.2). Pour évaluer si une association est marquante, nous avons utilisé une valeur seuil de FPRP de 0.2, comme suggéré par les auteurs12 dans les analyses récapitulatives. Par conséquent, les valeurs de FPRP de moins de 0.2 indiquent une association qui est demeurée robuste pour une probabilité antérieure donnée et désignée comme notable dans l'article actuel. La puissance statistique et les FPRP ont été calculées par le la feuille de calcul Excel fournie par Wacholder et coll. 12 égal à la valeur de P observée. 12 Mais compte tenu de l'attention récente portée à des OR beaucoup plus bas cette évaluation peut être trop conservatrice ; aussi, nous avons choisi de présenter les résultats pour des OR de 1.5 et 1.2 (ou leur réciproque 0.83=1/1.2). Pour évaluer si une association est marquante, nous avons utilisé une valeur seuil de FPRP de 0.2, comme suggéré par les auteurs12 dans les analyses récapitulatives. Par conséquent, les valeurs de FPRP de moins de 0.2 indiquent une association qui est demeurée robuste pour une probabilité antérieure donnée et désignée comme notable dans l'article actuel. La puissance statistique et les FPRP ont été calculées par le la feuille de calcul Excel fournie par Wacholder et coll. 12
Les gènes (National Center for Biotechnology Information Entrez identification numbers) dont les variants ont une association notable avec le cancer sont comme les suivants : MDM2 (4193), XPD (le nom a changé en ERCC2) (2068), RNASEL (6041), GSTT1 (2952) XRCC1 (7515), TGFB1 (7040), CASP8 (841), NAT2 (10), MTHFR (4524), CHEK2 (11200), et GSTM1 (2944).
RESULTATS
Nous avons identifié 161 méta-analyses et analyses groupées publiées, comprenant 18 localisations de cancer et 99 gènes différents. Ces 161 méta-analyses et analyses groupées ont traité de 344 associations entre cancer et variant de gène avec une moyenne de 7.3 études et 3551 cas par association étudiée (extrêmes, 508-19729 cas). Comme prévu, la plupart des analyses ont été conduites pour des cancers communs, tels que le sein (n=119), la prostate (n=42), et le cancer du poumon (n=34) ; il y avait très peu d'évaluations d’associations génétiques dans les cancers rares, tels que le cancer du col utérin et œsophagien (TABLEAU 1). Pour tous les sites de cancer, les variants des gènes impliqués dans la réparation d'ADN (par exemple, XRCC1 et XPD ; n=81) et les gènes codant pour le métabolisme enzymatique (par exemple, variants du cytochrome P450 (CYP), n=58 ; ou des S-transférases du glutathion (GST), n=31) ont le plus souvent été évaluées. Les méta-analyses et les analyses groupées ayant trouvé une association statistiquement significative ont évalué un nombre plus élevé d'études, mais ont inclus un nombre plus faible de cas que celles ayant trouvé une association non significative (P=0.02 et P=0.05, respectivement ; TABLEAU 1). A tableau complet présentant toutes les données extraites de chacune des 344 associations identifiées par notre recherche est disponible sur demande.
|
|
|
Tableau 1. Signification des associations variant génétique-cancer démontrée par méta-analyses et analyses groupées selon le site du cancer a
|
|
|
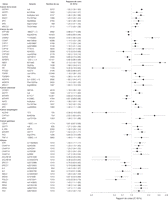
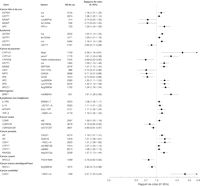
Dans les 344 associations cancer-variant de gène évaluées, les OR récapitulatifs pour les 98 associations (28%) (à l'exclusion de ceux impliquant HRAS1) étaient statistiquement significatif (valeurs de P entre 0.05 et 8.6 x 10–15 ; FIGURE 2 et FIGURE 3 et TABLEAU 2, TABLEAU 3, TABLEAU 4, et TABLEAU 5). Trente de ces 98 associations étaient inverses pour le variant, avec un OR moyen de 0.73 (médiane, 0.75 ; extrêmes, 0.32-0.92). Les 68 autres analyses ont rapporté des Or supérieurs à 1.0, avec une moyenne de 1.47 (médiane, 1.34 ; extrêmes, 1.07-3.13). Des associations statistiquement significatives ont été observées pour 16 sites de cancer, principalement dans les études du cancer du sein, du gliome, et du cancer du poumon.
|
|
|
Figure 2. Risque de cancer de la vessie, du sein, de l’œsophage et gastrique et de gliome selon les variants génétiques—limité aux méta-analyses et aux analyses groupées avec les estimations significatives de risque récapitulatif.
IC correspond à intervalle de confiance à 95%
|
|
|
|
|
|
Figure 3. Risque de cancer de la tête et du cou, poumon, ovarien, prostate, peau, tractus digestif supérieur, urothélial et leucémie, méningiome, et lymphome non Hodgkinien selon le variant génétique limité aux méta-analyses et aux analyses groupée avec des évaluations récapitulatives significatives du risque.
IC correspond à intervalle de confiance à 95%
|
|
|
|
|
|
Tableau 2. Associations statistiquement significatives cancer-variant génétique et probabilités de rapports faussement positifs pour le cancer du sein et le cancer colorectal
|
|
|
|
|
|
Tableau 3. Associations statistiquement significatives variant génétique-cancer et probabilités de rapports faussement positifs pour le cancer de la vessie, œsophagien, gastrique, et de la tête et du cou.
|
|
|
|
|
|
Tableau 4. Associations statistiquement significatives variant génétique-cancer et probabilités de rapport faussement positifs pour les cancers du poumon, cutanés non mélanome, ovarien, prostate, du tractus digestif supérieur, et urothélial.
|
|
|
|
|
|
Tableau 5. Associations statistiquement significatives variant génétique-cancer et probabilités de rapport faussement positifs pour le Gliome, la leucémie aiguë, le méningiome, et le lymphome non Hodgkinien
|
|
|
Pour évaluer la robustesse de ces résultats, nous avons calculé les valeurs de FPRP à 2 niveaux de probabilités antérieures. Pour les 98 associations, 85 associations cancer-variant de gène avaient des valeurs de FPRP supérieures à 0.2 dans les probabilités antérieures pré-spécifiées (0.001 et 0.000001) ; ces résultats ne sont pas considérés comme notables. Par exemple, bien que l’OR récapitulatif de l'analyse groupée pour XRCC1, Arg399Gln ait indiqué une association positive statistiquement significative avec un risque de cancer du sein (OR, 1.6 ; IC 95%, 1.1-2.3), les valeurs de FPRP étaient supérieures à 0.2, aux 2 probabilités antérieures; par conséquent, la conclusion n'est pas considérée comme notable. A un niveau de probabilité antérieure de 0.001 et pour une puissance statistique permettant de détecter un OR de 1.5, 13 associations cancer-variant de gène sont demeurées notables (FPRP  0.2) pour MDM2 SNP309 et cancer de poumon (OR, 1.27 ; P=0.0002) 55 ; XPD Lys751Gln et cancer du poumon (OR, 1.30 ; P=0.0002) 58; RNASEL Asp541Glu et cancer de la prostate (OR, 1.27 ; P=0.0001) 64; GSTT1 nul et cancer colorectal (OR, 1.37 ; P=8.1 x 10–5) 28 ; XRCC1 Arg399Gln et cancer de poumon (OR, 1.34 ; P=5.2 x 10–5) 59 ; TGFB1 Leu10Pro et cancer du sein (OR, 1.16 ; P = 6.9 x 10–5) 14 ; CASP8 Asp302His et cancer du sein (OR, 0.89 ; P=5.7 x 10–6) 14 ; NAT2 acétyleur lent et cancer de la vessie (OR, 1.46 ; P=2.5 x 10–7) 36 ; MTHFRC677T et cancer gastrique (OR, 1.52 ; P=4.9 x 10–8) 45 ; CHEK2 *1100delC et cancer du sein (OR, 2.4 ; P=2.5 x 10–9) 15 ; GSTT1 nul et leucémie aiguë (OR, 1.19 ; P=3.5 x 10–8) 68 ; GSTM1 nul et cancer de la vessie (OR, 1.5 ; P=1.9 x 10–14) 34 ; et GSTM1 nul et leucémie aiguë (OR, 1.20 ; P =8.6 x 10–15) 68 À une probabilité antérieure très basse de 0.000001, 4 de ces 13 associations cancer-variant de gène sont restés notables: MTHFR C677T, NAT2 acétyleur lent, et GSTM1 nul. Ce nombre s’est encore réduit à 2 (GSTM1 nul avec le cancer de la vessie et GSTM1 nul avec la leucémie) lorsque nous avons calculé la puissance statistique en se basant sur un OR inférieur à 1.2. Compatible avec la FPRP, les associations notables pour une probabilité antérieure très basse étaient fortement statistiquement significatives (valeurs de P entre 10–7 et 10–15). 0.2) pour MDM2 SNP309 et cancer de poumon (OR, 1.27 ; P=0.0002) 55 ; XPD Lys751Gln et cancer du poumon (OR, 1.30 ; P=0.0002) 58; RNASEL Asp541Glu et cancer de la prostate (OR, 1.27 ; P=0.0001) 64; GSTT1 nul et cancer colorectal (OR, 1.37 ; P=8.1 x 10–5) 28 ; XRCC1 Arg399Gln et cancer de poumon (OR, 1.34 ; P=5.2 x 10–5) 59 ; TGFB1 Leu10Pro et cancer du sein (OR, 1.16 ; P = 6.9 x 10–5) 14 ; CASP8 Asp302His et cancer du sein (OR, 0.89 ; P=5.7 x 10–6) 14 ; NAT2 acétyleur lent et cancer de la vessie (OR, 1.46 ; P=2.5 x 10–7) 36 ; MTHFRC677T et cancer gastrique (OR, 1.52 ; P=4.9 x 10–8) 45 ; CHEK2 *1100delC et cancer du sein (OR, 2.4 ; P=2.5 x 10–9) 15 ; GSTT1 nul et leucémie aiguë (OR, 1.19 ; P=3.5 x 10–8) 68 ; GSTM1 nul et cancer de la vessie (OR, 1.5 ; P=1.9 x 10–14) 34 ; et GSTM1 nul et leucémie aiguë (OR, 1.20 ; P =8.6 x 10–15) 68 À une probabilité antérieure très basse de 0.000001, 4 de ces 13 associations cancer-variant de gène sont restés notables: MTHFR C677T, NAT2 acétyleur lent, et GSTM1 nul. Ce nombre s’est encore réduit à 2 (GSTM1 nul avec le cancer de la vessie et GSTM1 nul avec la leucémie) lorsque nous avons calculé la puissance statistique en se basant sur un OR inférieur à 1.2. Compatible avec la FPRP, les associations notables pour une probabilité antérieure très basse étaient fortement statistiquement significatives (valeurs de P entre 10–7 et 10–15).
COMMENTAIRE
Globalement, près d'un tiers de toutes les associations cancer-variant de gène des méta-analyses et des analyses groupées publiées ont été rapportées comme étant statistiquement significatives. Treize de ces associations étaient notables pour une probabilité antérieure de 0.001 et une puissance statistique permettant de détecter un OR de 1.5, dont 4 sont restées notables même pour une probabilité antérieure inférieure semblable à une appropriée pour un SNP aléatoirement choisi dans une étude d'association génomique (1/1 000 000=0.000001) avec des valeurs de P entre 10–7 et 10–15. Ces associations ont ainsi moins de probabilité d’être des faux positifs et ont une probabilité élevée d'être de véritables associations de risque de cancer. Spécifiquement, nous avons observé que, parmi les associations notables, les gènes codant pour les enzymes du métabolisme de phase II ont composé la majorité des associations notables.
Les progrès continus des technologies de génotypage ont rendu possible l’examen d’un grand nombre de variants génétiques ; puis est venu la possibilité de publier un grand nombre de résultats faussement positifs en raison de la stratégie employée couramment de déclarer une signification sur une valeur de P de <0.05. Un dispositif principal de l'approche bayésienne utilisant la FPRP est qu'elle est basée, non seulement sur la valeur observée de P mais également sur la puissance et la probabilité antérieure de l'hypothèse, permettant à l'utilisateur d'incorporer les connaissances antérieures, y compris l'information fonctionnelle, des variants spécifiquement examinées. Bien que le calcul de FPRP permette une évaluation avec différents scénarios de probabilité antérieure, de puissance statistique, et du critère d'importance, le choix de ces paramètres devrait être déterminé a priori en utilisant les preuves empiriques des études passées. En conséquence, il peut être raisonnable de dire que les SNP des gènes candidats appropriés avec une fonction connue ou prévue (basée sur les études expérimentales ou les essais in silico) ont plus de probabilité d’être associées à un risque de cancer et par conséquent de justifier des probabilités antérieures plus élevées. Cependant, le choix d'une seule probabilité antérieure est sujet à discussion ; par conséquent, ici, nous fournissons aux lecteurs l'occasion d'employer leur propre jugement au sujet des preuves pour un gène candidat ou un variant donné. Dans cet article, nous avons choisi une approche plus agnostique aux associations d’évaluation en appliquant 2 niveaux de probabilité antérieure (0.001 et 0.000001) et de puissance statistique (OR 1.5, recommandé par Wacholder et coll. et semblable au OR médian rapporté dans notre revue) ; de même qu'à l’OR de 1.2, proche de l’OR médian rapporté dans notre revue) à toutes les associations statistiquement significatives. Comme l’ont suggéré Thomas et Clayton,72 la probabilité antérieure pour des études évaluant des gènes de candidat dépasse habituellement 1000:1 (ou 0.001). Aussi, pour une probabilité antérieure de 0.001, 13 associations étaient notables et peuvent plausiblement être de véritables associations. La probabilité d'être une association réelle est cependant encore plus élevée pour les 4 associations qui demeurent notables à une probabilité antérieure très basse (0.000001).
GSTM1 et GSTT1 appartiennent à une famille d’enzymes de phase II, les S-transférases du glutathion, qui sont impliquées dans le métabolisme et la métabolisation de xénobiotiques et d'endobiotiques toxiques. La suppression 73 de GSTT1 a été associée à un risque plus élevé de cancer 28 colorectal et de leucémie aiguë 68 et la délétion de GSTM1 est statistiquement significativement associée à un risque de cancer de la vessie 34 et de leucémie aiguë 68; et les 2 dernières se sont avérées être parmi les résultats les plus notables dans toutes les méta-analyses et analyses groupées. Les différentes études entreprises à la suite des méta-analyses continuent à soutenir les observations de GSTT174-79 et GSTM1, 80-85 sauf une étude qui indiquait une association inverse statistiquement significative entre GSTT1 nul et le cancer colorectal 86 et quelques petites études sur GSTT1 et leucémie fournissant des résultats inconsistants.83,85,87,88 La prévalence de GSTT1 nul va de 20% chez les blancs à 60% chez les Asiatiques, 89 et approximativement 50% des humains (allant de 22% en Afrique à 62% en Europe) ont GSTM1 nul.90 GSTT1 et GSTM1 sont impliqués dans l'élimination des carcinogènes dans l’organisme, tel que les produits du stress oxydant et les hydrocarbures aromatiques polycycliques de la fumée du tabac. 91 La délétion des gènes GSTT1 et GSTM1 entraîne dans le variant appelé GSTT1/GSTM1 nul une perte complète d'activité enzymatique.92 On s'attend ainsi à ce qu'un individu ayant des variants nuls ait une capacité altérée de détoxifier les carcinogènes et un plus grand risque de cancer, affectant potentiellement de multiples sites de cancer. Ceci et le fait que GSTT1 et GSTM1 entraînent des associations notables avec un risque de divers cancers soutient la théorie que ces 2 variants, en particulier GSTM1 sont fonctionnels et ont un impact véritable sur le risque de cancer.
Une autre observation parmi les plus notables a été l'association entre le phénotype NAT2 acétyleur lent et le cancer de la vessie.36 Cette méta-analyse a été publiée récemment ; aucune autre étude n'a été ainsi identifiée à la suite de la méta-analyse. NAT2 est 1 des 2 isoformes de la N-acétyl-transférase exprimés chez l'homme et impliqués dans la désintoxication des amines hétérocycliques ou aromatiques et de leur métabolites.93 NAT2 est fortement polymorphe et plusieurs polymorphismes non synonymes ont comme conséquence une faible expression, une protéine instable, ou une activité catalytique diminuée, qui ont comme conséquence un phénotype d’acétyleur lent.94 La prédominance de NAT2 acétyleurs lents chez les blancs européens est d’environ 56% et d’environ 11% chez les Asiatiques.34 La modification du taux d'acétylation pourrait altérer l'effet des carcinogènes sur le risque de cancer, mais l'impact de ce changement peut différer selon les site des cancers. Le phénotype NAT2 acétyleur lent est associé à un risque plus élevé de cancer de la vessie (dû à la diminution de la désintoxication des carcinogènes de la fumée de tabac), mais a été associé à une diminution du risque de cancer colorectal (dû à l'activation réduite des carcinogènes).31,93,94 Envisagées ensembles, les preuves irréfutables en faveur d’un effet fonctionnel du NAT2 acétyleur lent et l'association forte statistiquement significative avec le cancer de la vessie soutient l'hypothèse que cet variant est susceptible de modifier le risque de cancer.
L'association récemment publiée entre MTHFR C677T et le cancer gastrique était également parmi les associations les plus notables.45 MTHFR, la réductase du 5,10-méthyletetétrahydrofolate, joue un rôle principal dans la voie du métabolisme du 1-carbone. Spécifiquement, MTHFR convertit le 5,10-méthylenetétrahydrofolate en 5-méthyltétrahydrofolate qui permet alors le métabolisme de l'homocystéine et la fourniture de groupes méthyles. L'activité enzymatique chez les individus homozygotes pour MTHFRC677T est très réduite, approximativement à 30% de l’activité enzymatique prévue, comparés à ceux homozygotes pour le variant commun.95,96 En conséquence, la diminution de la capacité de MTHFR a été associée à des altérations des profils de méthylation et à une synthèse potentiellement anormale de l’ADN, de la réparation et de l’instabilité chromosomique.97 En raison de son rôle dans la voie principale, le variant de MTHFR C677T peut avoir un impact réel sur le risque de cancer.
Parmi les associations notables à la probabilité antérieure de 0.001 il y avait 3 gènes liés à la réparation d'ADN (CHEK2, XPD, et XRCC1). Les voies impliquant ces gènes sont responsables de la réparation des lésions et des erreurs de l’ADN pouvant se produire lors de la réplique de l’ADN. Il n'y a eu aucune étude publiée à la suite de la méta-analyse sur CHEK2 *1100delC et cancer du sein.15 Les études entreprises à la suite de la méta-analyse sur XPD Lys751Gln et cancer du poumon 98,99 ont tiré les mêmes conclusions que notre revue. Une conclusion statistiquement significative pour XRCC1 était présente chez les Asiatiques seulement, et 1 des 3 études suivantes entreprises chez les Asiatiques 100-102 a trouvé une association statistiquement significative entre XRCC1 Arg399Gln et cancer du poumon. De façon générale, il est biologiquement plausible que les gènes liés à la réparation de l’ADN aient un impact sur le risque de cancer et notre revue est en faveur de la probabilité de ces associations.
RNASEL Asp541Glu, MDM2 SNP309, TGFB1 Leu10Pro, et CASP8 Asp302His sont d’autres variants identifiés par notre revue en tant que notables; ils appartiennent aux voies principales influençant de façon plausible une susceptibilité au cancer. RNASEL joue un rôle important dans la voie de la réponse inflammatoire et a été identifié pour la première fois comme gène candidat du risque de cancer de la prostate en raison de sa localisation dans la région du cancer héréditaire 1 de la prostate (HPC1).103,104 Puisque une méta-analyse a été récemment publiée, seules 3 études publiées plus tard ont été identifiées avec des résultats contradictoires concernant le cancer de la prostate.105-107 MDM2 codes pour l'homologue humain de la souris double minute 2, une phospholipoprotéine nucléaire qui lie et inhibe p53, un suppresseur tumoral.108 Une autre étude publiée après la méta-analyse apporte un soutien lorsque l'analyse n'était pas limitée aux fumeurs.109 TGFB1, qui code le transforming growth factor beta 1, a été impliqué en tant que suppresseur tumoral et promoteur tumoral. 110,111 Une autre étude publiée ultérieurement n'a pas trouvé d’association.112 CASP8 code pour la caspase 8 qui joue un rôle central dans le déclenchement et l'activation d'une cascade de caspases menant à l'apoptose. 113 La diminution du risque avec CASP8 Asp302His de cancer du sein observé dans l'analyse groupée est encore appuyée par les résultats d'une étude récente d'association. 114
Très récemment, les résultats des premières études d'association génomique avec le cancer sont devenus disponibles, dans lesquelles des centaines de milliers de variants ont été génotypés dans le génome entier. Ces études ont détecté plusieurs variants fortement statistiquement significatifs dans la région du chromosome humain 8q24 associés au cancer de la prostate, colorectal, et à la prédisposition au cancer du sein ; cependant, il n'y a aucun gène caractérisé connu dans cette région.115-122 Les variants dans SMAD7,121 un gène impliqué dans les signaux cellulaires, et DAB2IP, 123 un gène suppresseur putatif tumoral, ont été également respectivement associés au cancer colorectal et de la prostate. Trois dépistages de suivi du génome dans le cancer de la prostate ont confirmé les loci précédemment identifiés et ont identifié plusieurs autres loci pouvant être associés au risque de cancer de la prostate.124-126 Les loci qui ont été identifiés dans au moins 2 des études étaient les suivants : 8q24, HNF1B (17q12), MSMB (10q11), NUDT10/11 (Xp11.22), et 17q24. Six variants fortement significatifs statistiquement étaient associés à une prédisposition au cancer du sein et ont été également identifiées par des études génomiques, dont 3 sont situés dans des gènes liés au contrôle de la croissance cellulaire ou du signal cellulaire (TNRC9, MAP3K1, et LSP1).122, 127,128 Deux variants étaient situés dans les régions 8q24 et 2q35, et le sixième dans FGFR2, un gène suppresseur des tumeurs surexprimé dans le cancer du sein. Des preuves substantielles soutenant ces variants, y compris la puissance importante et la réplication dans de grands échantillons, indiquent que ces associations sont susceptibles d'être réelles, mais aucune des variantes statistiquement significatives n'avait été précédemment identifiée car la plupart ne résidait pas dans des régions candidates « intéressantes ». Les études génomiques d'association au cancer ont également démontré que l’effet taille des variants génétiques statistiquement significatifs est tout à fait modeste globalement (point d’estimation entre 1.1 et 1.5 pour un mode additif de la transmission héréditaire), ce qui est compatible avec les faibles associations observées dans la plupart des méta-analyses et analyses groupées.
Nous avons essayé de passer en revue toutes les méta-analyses et les analyses groupées publiées couvrant le sujet des variants génétiques et risque de cancer par plusieurs itérations de critère de recherche ; il est cependant possible que nous ayons manqué quelques études. Plusieurs des variants notables identifiées étaient des délétions (qui peuvent ne pas être bien capturées par les études génomiques d'association) et des SNP non synonymes, mais ceci peut être dû au fait que ces types de mutations tendent à être le plus généralement étudiés. Notre objectif portait strictement sur les résultats des études d'association des gènes candidats et n'a pas pris en considération les résultats des études de linkage pour identifier les gènes à haute pénétrance. Une autre limite potentielle de cette revue est que les associations étaient confinées à celles récapitulées par une méta-analyse ou une analyse groupée. Nous connaissons différentes études ayant des dimensions d'échantillon potentiellement beaucoup plus élevées et par conséquent plus de puissance pour mettre en évidence une association statistiquement significative que certaines méta-analyses et analyses groupées ; certaines de ces études ont été entreprises à la suite de méta-analyses ou des analyses groupées et certaines avant. Pour répondre en partie à cette question, nous avons passé en revue des études entreprises à la suite des dernières méta-analyses ou d’analyses groupées pour des associations considérées comme notables avec une faible probabilité antérieure pour déterminer si les preuves continuaient à appuyer les associations précédemment observées. Une autre limite de notre revue est que nos résultats sont susceptibles de diminuer la qualité et la largeur des méta-analyses ou des analyses groupées en raison des biais de publication. Cependant, la plupart des analyses incluses ci-dessus ont évalué les biais et l'hétérogénéité des publications, comme indiqué dans les tableaux ci-joints. Puisque la puissance pour évaluer les interactions gène-gène et gène-environnement est inférieure à celle pour évaluer les effets principaux et puisque la plupart des méta-analyses et analyses groupées se concentraient sur les effets principaux, nous avons seulement rendu compte des effets principaux des variants génétiques. Par conséquent, nous pouvons avoir manqué d’importants effets dans des sous-groupes, car il est possible que certains variants génétiques ne puissent être appropriés que lorsque « le système est sous stress, » par exemple, tabagisme, maladie concourante, ou malnutrition. La plupart des analyses ont évalué les polymorphismes de candidats uniques ; mais, le génotypage étant devenu de plus en plus accessible ces dernières années, ceci permet maintenant aux investigateurs de déterminer les variants génétiques dans des gènes et des voies candidats entiers et plus récemment dans le génome entier. Bien qu'il soit facile de comparer les résultats des SNP simples, cette approche est certainement moins complète et n'élimine pas que d’autres SNP dans le même gène peut être liés à un risque de cancer. Comme le nombre d'articles sur les variants génétiques publiés au cours de la dernière décennie a augmenté considérablement et continue à se développer, nous acceptons que cette revue ne demeure pas longtemps d’actualité, mais fournissons un instantané des progrès dans ce domaine.
Nous avons observé 98 associations statistiquement significatives entre cancer et variant génétique, dont 13 ont été considérés notables pour une probabilité antérieure de 0.001. Avec une probabilité antérieure très basse (0.000001), 4 sont restés notables et toutes étaient fortement significatives sur le plan statistique (valeurs de P entre 10–7 et 10–15). Une majorité des associations les plus notables identifiées ne sont pas des SNP mais des délétions, 4 impliquent des variants de GST. Les résultats des méta-analyses et des analyses groupées ont été utiles pour synthétiser les résultats publiés et peuvent guider les futures études génétiques vers des secteurs qui exigent davantage de clarification et à s’éloigner de ceux qui n’en ont pas besoin.
Informations sur les auteurs
Correspondance: Ulrike Peters, PhD, MPH, Cancer Prevention Program (M4-B402), Fred Hutchinson Cancer Research Center, PO Box 19024, Seattle, WA 98109 (upeters{at}fhcrc.org)
Contributions des auteurs: Les Drs Dong et Peters ont un accès complet à toutes les données de l'étude et accepte la responsabilité de l'intégrité des données et de l'exactitude de l'analyse de données.
Conception et schéma de l’étude: Peters.
Recueil des données: Dong, Peters.
Analyse et interprétation des données: Dong, Potter, White, Ulrich, Cardon, Peters.
Rédaction du manuscrit: Dong, Peters.
Revue critique du manuscrit: Dong, Potter, White, Ulrich, Cardon, Peters.
Analyse statistique: Dong, Cardon, Peters.
Obtention du financement: White, Peters.
Aide administrative, technique, et matérielle: Potter, Peters.
Supervision de l’étude: White, Peters.
Liens financiers: Le Dr. Cardon a servi de conseiller à Illumnia. Aucune autre déclaration financière n'a été faite.
Financement/Soutien: Cette recherche a été soutenue en partie par des bourses R25 CA94880, CA118421, et CA059045 du National Institutes of Health (NIH).
Rôle du sponsor: Le commanditaire n'a joué aucun rôle dans la conception et la conduite de l'étude ; la collection, gestion, analyse, et interprétation des données ; ou préparation, revue, ou approbation du manuscrit.
Autres contributions: Nous remercions Rothman national, MD, MPH, MHS, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, Maryland), Institut National contre le Cancer, NIH, Bethesda, Maryland), pour ses commentaires utiles sur la FPRP. Il n'a pas reçu de compensation pour ses contributions.
Affiliations des auteurs: Fred Hutchinson Cancer Research Center, Seattle, Washington (Drs Dong, Potter, White, Ulrich, Cardon, et Peters) et Departments of Epidemiology (Drs Dong, Potter, White, Ulrich, et Peters) et Biostatistics (Dr Cardon), University of Washington, Seattle.
FMC disponible en ligne à www.jamaarchivescme.com et questions p 2454.
BIBLIOGRAPHIE
| |
1. Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome [published correction appears in Nature. 2001;412(6846):565]. Nature. 2001;409(6822):860-921.
PUBMED
2. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304-1351.
FREE FULL TEXT
3. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431(7011):931-945.
PUBMED
4. The International HapMap Consortium. The International HapMap Project. Nature. 2003;426(6968):789-796.
PUBMED
5. Lin BK, Clyne M, Walsh M, et al. Tracking the epidemiology of human genes in the literature: the HuGE Published Literature database. Am J Epidemiol. 2006;164(1):1-4.
FREE FULL TEXT
6. Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29(3):306-309.
PUBMED
7. Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA. 2007;297(14):1551-1561.
FREE FULL TEXT
8. Dickersin K, Berlin JA. Meta-analysis: state-of-the-science. Epidemiol Rev. 1992;14:154-176.
FREE FULL TEXT
9. Mosteller F, Colditz GA. Understanding research synthesis (meta-analysis). Annu Rev Public Health. 1996;17:1-23.
PUBMED
10. Morris RD. Meta-analysis in cancer epidemiology. Environ Health Perspect. 1994;102(suppl 8):61-66.
11. Poole C.. Low P-values or narrow confidence intervals: which are more durable? Epidemiology. 2001;12(3):291-294.
PUBMED
12. Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96(6):434-442.
FREE FULL TEXT
13. Garcia-Closas M, Kristensen V, Langerød A, et al. Common genetic variation in TP53 and its flanking genes, WDR79 and ATP1B2, and susceptibility to breast cancer. Int J Cancer. 2007;121(11):2532-2538.
PUBMED
14. Cox A, Dunning AM, Garcia-Closas M, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39(3):352-358.
PUBMED
15. Weischer M, Bojesen SE, Ellervik C, Tybjaerg-Hansen A, Nordestgaard BG. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol. 2008;26(4):542-548.
FREE FULL TEXT
16. Onay UV, Aaltonen K, Briollais L, et al. Combined effect of CCND1 andCOMTpolymorphisms and increased breast cancer risk. BMC Cancer. 2008;8:6.
PUBMED
17. Setiawan VW, Schumacher FR, Haiman CA, et al. CYP17 genetic variation and risk of breast and prostate cancer from the National Cancer Institute Breast and Prostate Cancer Cohort Consortium (BPC3). Cancer Epidemiol Biomarkers Prev. 2007;16(11):2237-2246.
FREE FULL TEXT
18. de Jong MM, Nolte IM, te Meerman GJ, et al. Genes other than BRCA1 and BRCA2 involved in breast cancer susceptibility. J Med Genet. 2002;39(4):225-242.
FREE FULL TEXT
19. Chen C, Huang Y, Li Y, Mao Y, Xie Y. Cytochrome P450 1A1 (CYP1A1) T3801C and A2455G polymorphisms in breast cancer risk: a meta-analysis. J Hum Genet. 2007;52(5):423-435.
PUBMED
20. Paracchini V, Raimondi S, Gram IT, et al. Metaand pooled analyses of the cytochrome P-450 1B1 Val432Leu polymorphism and breast cancer: a HuGE-GSEC review. Am J Epidemiol. 2007;165(2):115-125.
FREE FULL TEXT
21. Garcia-Closas M, Troester MA, Qi Y, et al. Common genetic variation in GATA-binding protein 3 and differential susceptibility to breast cancer by estrogen receptor alpha tumor status. Cancer Epidemiol Biomarkers Prev. 2007;16(11):2269-2275.
FREE FULL TEXT
22. Steffen J, Nowakowska D, Niwinska A, et al. Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int J Cancer. 2006;119(2):472-475.
PUBMED
23. Haiman CA, Setiawan VW, Xia LY, et al. A variant in the cytochrome p450 oxidoreductase gene is associatedwith breast cancer risk in African Americans. Cancer Res. 2007;67(8):3565-3568.
FREE FULL TEXT
24. Cox DG, Buring J, Hankinson SE, Hunter DJ. A polymorphism in the 3 untranslated region of the gene encoding prostaglandin endoperoxide synthase 2 is not associated with an increase in breast cancer risk: a nested case-control study. Breast Cancer Res. 2007;9(1):R3.
PUBMED
25. Pasche B, Kaklamani V, Hou N, et al. TGFBR1*6A and cancer: ameta-analysis of 12 case-control studies. J Clin Oncol. 2004;22(4):756-758.
FREE FULL TEXT
26. Zhang Y, Newcomb PA, Egan KM, et al. Genetic polymorphisms in base-excision repair pathway genes and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(2):353-358.
FREE FULL TEXT
27. Tan XL, Nieters A, Kropp S, Hoffmeister M, Brenner H, Chang-Claude J. The association of cyclin D1 G870A and E-cadherin C-160A polymorphisms with the risk of colorectal cancer in a case control study and meta-analysis. Int J Cancer. 2008;122(11):2573-2580.
PUBMED
28. de Jong MM, Nolte IM, te Meerman GJ, et al. Lowpenetrance genes and their involvement in colorectal cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 2002;11(11):1332-1352.
FREE FULL TEXT
29. Hubner RA, Houlston RS. MTHFR C677T and colorectal cancer risk: a meta-analysis of 25 populations. Int J Cancer. 2007;120(5):1027-1035.
PUBMED
30. Huang Y, Han S, Li Y, Mao Y, Xie Y. Different roles of MTHFR C677T and A1298C polymorphisms in colorectal adenoma and colorectal cancer: a meta-analysis. J Hum Genet. 2007;52(1):73-85.
PUBMED
31. Chen K, Jiang QT, He HQ. Relationship between metabolic enzyme polymorphism and colorectal cancer. World J Gastroenterol. 2005;11(3):331-335.
PUBMED
32. Chao C, Zhang ZF, Berthiller J, Boffetta P, Hashibe M. NAD(P)H:quinone oxidoreductase 1 (NQO1) Pro187Ser polymorphism and the risk of lung, bladder, and colorectal cancers: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15(5):979-987.
FREE FULL TEXT
33. Zhang D, Chen C, Fu X, et al. A meta-analysis of DNA repair gene XPC polymorphisms and cancer risk. J Hum Genet. 2008;53(1):18-33.
PUBMED
34. Garc untranslated region of the gene encoding prostaglandin endoperoxide synthase 2 is not associated with an increase in breast cancer risk: a nested case-control study. Breast Cancer Res. 2007;9(1):R3.
PUBMED
25. Pasche B, Kaklamani V, Hou N, et al. TGFBR1*6A and cancer: ameta-analysis of 12 case-control studies. J Clin Oncol. 2004;22(4):756-758.
FREE FULL TEXT
26. Zhang Y, Newcomb PA, Egan KM, et al. Genetic polymorphisms in base-excision repair pathway genes and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(2):353-358.
FREE FULL TEXT
27. Tan XL, Nieters A, Kropp S, Hoffmeister M, Brenner H, Chang-Claude J. The association of cyclin D1 G870A and E-cadherin C-160A polymorphisms with the risk of colorectal cancer in a case control study and meta-analysis. Int J Cancer. 2008;122(11):2573-2580.
PUBMED
28. de Jong MM, Nolte IM, te Meerman GJ, et al. Lowpenetrance genes and their involvement in colorectal cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 2002;11(11):1332-1352.
FREE FULL TEXT
29. Hubner RA, Houlston RS. MTHFR C677T and colorectal cancer risk: a meta-analysis of 25 populations. Int J Cancer. 2007;120(5):1027-1035.
PUBMED
30. Huang Y, Han S, Li Y, Mao Y, Xie Y. Different roles of MTHFR C677T and A1298C polymorphisms in colorectal adenoma and colorectal cancer: a meta-analysis. J Hum Genet. 2007;52(1):73-85.
PUBMED
31. Chen K, Jiang QT, He HQ. Relationship between metabolic enzyme polymorphism and colorectal cancer. World J Gastroenterol. 2005;11(3):331-335.
PUBMED
32. Chao C, Zhang ZF, Berthiller J, Boffetta P, Hashibe M. NAD(P)H:quinone oxidoreductase 1 (NQO1) Pro187Ser polymorphism and the risk of lung, bladder, and colorectal cancers: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15(5):979-987.
FREE FULL TEXT
33. Zhang D, Chen C, Fu X, et al. A meta-analysis of DNA repair gene XPC polymorphisms and cancer risk. J Hum Genet. 2008;53(1):18-33.
PUBMED
34. Garc ’a-Closas M. Malats N, Silverman D, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366(9486):649-659.
PUBMED
35. Kellen E, Hemelt M, Broberg K, et al. Pooled analysis and meta-analysis of the glutathione Stransferase P1 Ile 105Val polymorphism and bladder cancer: a HuGE-GSEC review. Am J Epidemiol. 2007;165(11):1221-1230.
FREE FULL TEXT
36. Sanderson S, Salanti G, Higgins J. Joint effects of the N-Acetyltransferase 1 and 2 (NAT1 and NAT2) genes and smoking on bladder carcinogenesis: a literature- based Systematic HuGE review and evidence synthesis. Am J Epidemiol. 2007;166(7):741.
FREE FULL TEXT
37. Francisco G, Menezes PR, Eluf-Neto J, Chammas R. XPC polymorphisms play a role in tissue-specific carcinogenesis: a meta-analysis [published online February 20, 2008]. Eur J Hum Genet. doi:10.1038/ejhg.2008.6.38. Wang F, Chang D, Hu FL, et al. DNA repair gene XPD polymorphisms and cancer risk: a meta-analysis based on 56 case-control studies. Cancer Epidemiol Biomarkers Prev. 2008;17(3):507-517.
FREE FULL TEXT
39. Figueroa JD, Malats N, Rothman N, et al. Evaluation of genetic variation in the double-strand break repair pathway and bladder cancer risk. Carcinogenesis. 2007;28(8):1788-1793.
FREE FULL TEXT
40. Lewis SJ, Smith GD. Alcohol, ALDH2, and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1967-1971.
FREE FULL TEXT
41. Yang CX, Matsuo K, Wang ZM, Tajima K. Phase I/II enzyme gene polymorphisms and esophageal cancer risk: a meta-analysis of the literature. World J Gastroenterol. 2005;11(17):2531-2538.
PUBMED
42. Wang GY, Lu CQ, Zhang RM, Hu XH, Luo ZW. The E-cadherin gene polymorphism 160C-A and cancer risk: A HuGE review and meta-analysis of 26 case-control studies. Am J Epidemiol. 2008;167(1):7-14.
FREE FULL TEXT
43. Boccia S, La Torre G, Gianfagna F, Mannocci A, Ricciardi G. Glutathione S-transferase T1 status and gastric cancer risk: a meta-analysis of the literature. Mutagenesis. 2006;21(2):115-123.
FREE FULL TEXT
44. Wang P, Xia HH, Zhang JY, Dai LP, Xu XQ, Wang KJ. Association of interleukin-1 gene polymorphisms with gastric cancer: ameta-analysis. Int J Cancer. 2007;120(3):552-562.
PUBMED
45. Boccia S, Hung R, Ricciardi G, et al. Meta- and pooled analyses of the methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and gastric cancer risk: a huge-GSEC review. Am J Epidemiol. 2008;(5):505-516.46. Zhou Y, Li N, Zhuang W, et al. P53 codon 72 polymorphism and gastric cancer: a meta-analysis of the literature. Int J Cancer. 2007;121(7):1481-1486.
PUBMED
47. Gorouhi F, Islami F, Bahrami H, Kamangar F. Tumour-necrosis factor-A polymorphisms and gastric cancer risk: a meta-analysis [published online March 4, 2008]. Br J Cancer. doi:10.1038/sj.bjc.6604277.48. Tripathy CB, Roy N. Meta-analysis of glutathione S-transferase M1 genotype and risk toward head and neck cancer. Head Neck. 2006;28(3):217-224.
PUBMED
49. Ye Z, Song H, Guo Y. Glutathione S-transferase M1, T1 status and the risk of head and neck cancer: a meta-analysis. J Med Genet. 2004;41(5):360-365.
FREE FULL TEXT
50. Huang WY, Olshan AF, Schwartz SM, et al. Selected genetic polymorphisms inMGMT, XRCC1, XPD, and XRCC3 and risk of head and neck cancer: a pooled analysis. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1747-1753.
FREE FULL TEXT
51. Vineis P, Veglia F, Benhamou S, et al. CYP1A1 T3801 C polymorphism and lung cancer: a pooled analysis of 2451 cases and 3358 controls. Int J Cancer. 2003;104(5):650-657.
PUBMED
52. Shi X, Zhou S, Wang Z, Zhou Z, Wang Z. CYP1A1 and GSTM1 polymorphisms and lung cancer risk in Chinese populations: a meta-analysis. Lung Cancer. 2008;59(2):155-163.
PUBMED
53. Rostami-Hodjegan A, Lennard MS, Woods HF, Tucker GT. Meta-analysis of studies of the CYP2D6 polymorphism in relation to lung cancer and Parkinson’s disease. Pharmacogenetics. 1998;8(3):227-238.
PUBMED
54. Raimondi S, Paracchini V, Autrup H, et al. Metaand pooled analysis of GSTT1 and lung cancer: a HuGE-GSEC review. Am J Epidemiol. 2006;164(11):1027-1042.
FREE FULL TEXT
55. Wilkening S, Bermejo JL, Hemminki K. MDM2 SNP309 and cancer risk: a combined analysis. Carcinogenesis. 2007;28(11):2262-2267.
FREE FULL TEXT
56. Lee WJ, Brennan P, Boffetta P, et al. Microsomal epoxide hydrolase polymorphisms and lung cancer risk: a quantitative review. Biomarkers. 2002;7(3):230-241.
PUBMED
57. Taioli E, Benhamou S, Bouchardy C, et al. Myeloperoxidase G-463A polymorphism and lung cancer: a HuGE genetic susceptibility to environmental carcinogens pooled analysis. Genet Med. 2007;9(2):67-73.
PUBMED
58. Kiyohara C, Yoshimasu K. Genetic polymorphisms in the nucleotide excision repair pathway and lung cancer risk: ameta-analysis. Int JMed Sci. 2007;4(2):59-71.
59. Kiyohara C, Takayama K, Nakanishi Y. Association of genetic polymorphisms in the base excision repair pathway with lung cancer risk: a meta-analysis. Lung Cancer. 2006;54(3):267-283.
PUBMED
60. Han S, Zhang HT, Wang Z, et al. DNA repair gene XRCC3 polymorphisms and cancer risk: a metaanalysis of 48 case-control studies. Eur J Hum Genet. 2006;14(10):1136-1144.
PUBMED
61. Rothman N, Skibola CF, Wang SS, et al. Genetic variation in TNF and IL10 and risk of non-Hodgkin lymphoma: a report from the InterLymph Consortium. Lancet Oncol. 2006;7(1):27-38.
PUBMED
62. Gayther SA, Song H, Ramus SJ, et al. Tagging single nucleotide polymorphisms in cell cycle control genes and susceptibility to invasive epithelial ovarian cancer. Cancer Res. 2007;67(7):3027-3035.
FREE FULL TEXT
63. Zeegers MP, Kiemeney LA, Nieder AM, Ostrer H. How strong is the association between CAG and GGN repeat length polymorphisms in the androgen receptor gene and prostate cancer risk? Cancer Epidemiol Biomarkers Prev. 2004;13(11 pt 1):1765-1771.
FREE FULL TEXT
64. Li H, Tai BC. RNASEL gene polymorphisms and the risk of prostate cancer: ameta-analysis. Clin Cancer Res. 2006;12(19):5713-5719.
FREE FULL TEXT
65. Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a HuGE Review. Am J Epidemiol. 2005;162(10):925-942.
FREE FULL TEXT
66. Bethke L, Webb E, Murray A, et al. Comprehensive analysis of the role of DNA repair gene polymorphisms on risk of glioma. Hum Mol Genet. 2008;17(6):800-805.
FREE FULL TEXT
67. Brenner AV, Butler MA, Wang SS, et al. Singlenucleotide polymorphisms in selected cytokine genes and risk of adult glioma. Carcinogenesis. 2007;28(12):2543-2547.
FREE FULL TEXT
68. Ye Z, Song H. Glutathione s-transferase polymorphisms (GSTM1, GSTP1 and GSTT1) and the risk of acute leukaemia: a systematic review and meta-analysis. Eur J Cancer. 2005;41(7):980-989.
PUBMED
69. Pereira TV, Rudnicki M, Pereira AC. Pombo-de- Oliveira MS, Franco RF. 5,10-Methylenetetrahydrofolate reductase polymorphisms and acute lymphoblastic leukemia risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1956-1963.
FREE FULL TEXT
70. Bethke L, Murray A, Webb E, et al. Comprehensive analysis of DNA repair gene variants and risk of meningioma. J Natl Cancer Inst. 2008;100(4):270-276.
FREE FULL TEXT
71. Lee KM, Lan Q, Kricker A, et al. One-carbon metabolism gene polymorphisms and risk of non- Hodgkin lymphoma in Australia. Hum Genet. 2007;122(5):525-533.
PUBMED
72. Thomas DC, Clayton DG. Betting odds and genetic associations. J Natl Cancer Inst. 2004;96(6):421-423.
FREE FULL TEXT
73. Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30(6):445-600.
PUBMED
74. Yeh CC, Sung FC, Tang R, Chang-Chieh CR, Hsieh LL. Association between polymorphisms of biotransformation and DNA-repair genes and risk of colorectal cancer in Taiwan. J Biomed Sci. 2007;14(2):183-193.
PUBMED
75. Mart’nez C. Mart’n F, Ferna’ndez JM, et al. Glutathione S-transferases mu 1, theta 1, pi 1, alpha 1 and mu 3 genetic polymorphisms and the risk of colorectal and gastric cancers in humans. Pharmacogenomics. 2006;7(5):711-718.
PUBMED
76. Little J, Sharp L, Masson LF, et al. Colorectal cancer and genetic polymorphisms of CYP1A1, GSTM1 and GSTT1: a case-control study in the Grampian region of Scotland. Int J Cancer. 2006;119(9):2155-2164.
PUBMED
77. Rajagopal R, Deakin M, Fawole AS, et al. Glutathione S-transferase T1 polymorphisms are associated with outcome in colorectal cancer. Carcinogenesis. 2005;26(12):2157-2163.
FREE FULL TEXT
78. Ates NA, Tamer L, Ates C, et al. Glutathione Stransferase M1, T1, P1 genotypes and risk for development of colorectal cancer. Biochem Genet. 2005;43(3-4):149-163.
PUBMED
79. Bolufer P, Collado M, Barragan E, et al. The potential effect of gender in combination with common genetic polymorphisms of drug-metabolizing enzymes on the risk of developing acute leukemia. Haematologica. 2007;92(3):308-314.
FREE FULL TEXT
80. Vineis P, Veglia F, Garte S, et al. Genetic susceptibility according to three metabolic pathways in cancers of the lung and bladder and in myeloid leukemias in nonsmokers. Ann Oncol. 2007;18(7):1230-1242.
FREE FULL TEXT
81. Cengiz M, Ozaydin A, Ozkilic AC, Dedekarginoglu G. The investigation of GSTT1, GSTM1 and SOD polymorphism in bladder cancer patients. Int Urol Nephrol. 2007;39(4):1043-1048.
PUBMED
82. Bolufer P, Barragan E, Collado M, Cervera J, Lopez JA, Sanz MA. Influence of genetic polymorphisms on the risk of developing leukemia and on disease progression. Leuk Res. 2006;30(12):1471-1491.
PUBMED
83. Aydin-SayitogluM. Hatirnaz O, Erensoy N, Ozbek U. Role of CYP2D6, CYP1A1, CYP2E1, GSTT1, and GSTM1 genes in the susceptibility to acute leukemias. Am J Hematol. 2006;81(3):162-170.
PUBMED
84. Shao J, Gu M, Zhang Z, Xu Z, Hu Q, Qian L. Genetic variants of the cytochrome P450 and glutathione S-transferase associated with risk of bladder cancer in a south-eastern Chinese population. Int J Urol. 2008;15(3):216-221.
PUBMED
85. Majumdar S, Mondal BC, Ghosh M, et al. Association of cytochrome P450, glutathione Stransferase and N-acetyl transferase 2 gene polymor- phisms with incidence of acute myeloid leukemia. Eur J Cancer Prev. 2008;17(2):125-132.
PUBMED
86. Huang K, Sandler RS, Millikan RC, Schroeder JC, North KE, Hu J. GSTM1 and GSTT1 polymorphisms, cigarette smoking, and risk of colon cancer: a population- based case-control study in North Carolina (United States). Cancer Causes Control. 2006;17(4):385-394.
PUBMED
87. Clavel J, Bellec S, Rebouissou S, et al. Childhood leukaemia, polymorphisms of metabolism enzyme genes, and interactions with maternal tobacco, coffee and alcohol consumption during pregnancy. Eur J Cancer Prev. 2005;14(6):531-540.
PUBMED
88. Pakakasama S, Mukda E, Sasanakul W, et al. Polymorphisms of drug-metabolizing enzymes and risk of childhood acute lymphoblastic leukemia. Am J Hematol. 2005;79(3):202-205.
PUBMED
89. Nelson HH, Wiencke JK, Christiani DC, et al. Ethnic differences in the prevalence of the homozygous deleted genotype of glutathione S-transferase theta. Carcinogenesis. 1995;16(5):1243-1245.
FREE FULL TEXT
90. Rebbeck TR. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 1997;6(9):733-743.
FREE FULL TEXT
91. Hayes JD, Strange RC. Potential contribution of the glutathione S-transferase supergene family to resistance to oxidative stress. Free Radic Res. 1995;22(3):193-207.
PUBMED
92. Parl FF. Glutathione S-transferase genotypes and cancer risk. Cancer Lett. 2005;221(2):123-129.
PUBMED
93. Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506-507:65-77.
94. Hein DW, Doll MA, Fretland AJ, et al. Molecular genetics and epidemiology of the NAT1 and NAT acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9(1):29-42.
FREE FULL TEXT
95. Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10(1):111-113.
PUBMED
96. Rozen R.. Genetic predisposition to hyperhomocysteinemia: deficiency of methylenetetrahydrofolate reductase (MTHFR). Thromb Haemost. 1997;78(1):523-526.
PUBMED
97. Choi SW, Mason JB. Folate status: effects on pathways of colorectal carcinogenesis. J Nutr. 2002;132(8)(suppl):2413S-2418S.
FREE FULL TEXT
98. Ló pez-Cima MF, Gonza lez-Arriaga P, Garca-Castro L, et al. Polymorphisms in XPC, XPD, XRCC1, and XRCC3 DNA repair genes and lung cancer risk in a population of Northern Spain.BMCCancer. 2007;7:162.99. De Ruyck K, Szaumkessel M, De Rudder I, et al. Polymorphisms in base-excision repair and nucleotideexcision repair genes in relation to lung cancer risk. Mutat Res. 2007;631(2):101-110.
PUBMED
100. Pachouri SS, Sobti RC, Kaur P, Singh J. Contrasting impact of DNA repair gene XRCC1 polymorphisms Arg399Gln and Arg194Trp on the risk of lung cancer in the north-Indian population. DNA Cell Biol. 2007;26(3):186-191.
PUBMED
101. Yin J, Vogel U, Ma Y, Qi R, Sun Z, Wang H. The DNA repair gene XRCC1 and genetic susceptibility of lung cancer in a northeastern Chinese population. Lung Cancer. 2007;56(2):153-160.
PUBMED
102. Sreeja L, Syamala VS, Syamala V, et al. Prognostic importance of DNA repair gene polymorphisms of XRCC1 Arg399Gln and XPD Lys751Gln in lung cancer patients from India. J Cancer Res Clin Oncol. 2008;134(6):645-652.
PUBMED
103. Sun J, Turner A, Xu J, Gronberg H, Isaacs W. Genetic variability in inflammation pathways and prostate cancer risk. Urol Oncol. 2007;25(3):250-259.
PUBMED
104. Smith JR, Freije D, Carpten JD, et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996;274(5291):1371-1374.
FREE FULL TEXT
105. Shook SJ, Beuten J, Torkko KC, et al. Association of RNASEL variants with prostate cancer risk in Hispanic Caucasians and African Americans. Clin Cancer Res. 2007;13(19):5959-5964.
FREE FULL TEXT
106. Cybulski C, Wokolorczyk D, Jakubowska A, et al. DNA variation in MSR1, RNASEL and E-cadherin genes and prostate cancer in Poland. Urol Int. 2007;79(1):44-49.
PUBMED
107. Shea PR, Ishwad CS, Bunker CH, Patrick AL, Kuller LH, Ferrell RE. RNASEL and RNASEL-inhibitor variation and prostate cancer risk in Afro-Caribbeans. Prostate. 2008;68(4):354-359.
PUBMED
108. Momand J, Wu HH, Dasgupta G. MDM2– master regulator of the p53 tumor suppressor protein. Gene. 2000;242(1-2):15-29.
PUBMED
109. Liu G, Wheatley-Price P, Zhou W, et al. Genetic polymorphisms of MDM2, cumulative cigarette smoking and nonsmall cell lung cancer risk. Int J Cancer. 2008;122(4):915-918.
PUBMED
110. Glick AB. TGFbeta1, back to the future: revisiting its role as a transforming growth factor. Cancer Biol Ther. 2004;3(3):276-283.
PUBMED
111. Rosfjord EC, Dickson RB. Growth factors, apoptosis, and survival of mammary epithelial cells.JMammary Gland Biol Neoplasia. 1999;4(2):229-237.
112. Cox DG, Penney K, Guo Q, Hankinson SE, Hunter DJ. TGFB1 and TGFBR1 polymorphisms and breast cancer risk in the Nurses’ Health Study. BMC Cancer. 2007;7:175.
PUBMED
113. Grenet J, Teitz T, Wei T, Valentine V, Kidd VJ. Structure and chromosome localization of the human CASP8 gene. Gene. 1999;226(2):225-232.
PUBMED
114. Sigurdson AJ, Bhatti P, Doody MM, et al. Polymorphisms in apoptosis- and proliferation-related genes, ionizing radiation exposure, and risk of breast cancer among US Radiologic Technologists. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2000-2007.
FREE FULL TEXT
115. Gudmundsson J, Sulem P, Manolescu A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39(5):631-637.
PUBMED
116. Yeager M, Orr N, Hayes RB, et al. Genomewide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39(5):645-649.
PUBMED
117. Haiman CA, Patterson N, Freedman ML, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39(5):638-644.
PUBMED
118. Tomlinson I, Webb E, Carvajal-Carmona L, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39(8):984-988.
PUBMED
119. Haiman CA, Le Marchand L, Yamamato J, et al. A common genetic risk factor for colorectal and prostate cancer. Nat Genet. 2007;39(8):954-956.
PUBMED
120. Zanke BW, Greenwood CM, Rangrej J, et al. Genome- wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39(8):989-994.
PUBMED
121. Broderick P, Carvajal-Carmona L, Pittman AM, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39(11):1315-1317.
PUBMED
122. Easton DF, Pooley KA, Dunning AM, et al. Genome- wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148): 1087-1093.
PUBMED
123. Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99(24):1836-1844.
FREE FULL TEXT
124. Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40(3):316-321.
PUBMED
125. Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40(3):310-315.
PUBMED
126. Gudmundsson J, Sulem P, Rafnar T, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40(3):281-283.
PUBMED
127. Hunter DJ, Kraft P, Jacobs KB, et al. A genomewide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39(7):870-874.
PUBMED
128. Stacey SN, Manolescu A, Sulem P, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39(7):865-869.
PUBMED ’a-Closas M. Malats N, Silverman D, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366(9486):649-659.
PUBMED
35. Kellen E, Hemelt M, Broberg K, et al. Pooled analysis and meta-analysis of the glutathione Stransferase P1 Ile 105Val polymorphism and bladder cancer: a HuGE-GSEC review. Am J Epidemiol. 2007;165(11):1221-1230.
FREE FULL TEXT
36. Sanderson S, Salanti G, Higgins J. Joint effects of the N-Acetyltransferase 1 and 2 (NAT1 and NAT2) genes and smoking on bladder carcinogenesis: a literature- based Systematic HuGE review and evidence synthesis. Am J Epidemiol. 2007;166(7):741.
FREE FULL TEXT
37. Francisco G, Menezes PR, Eluf-Neto J, Chammas R. XPC polymorphisms play a role in tissue-specific carcinogenesis: a meta-analysis [published online February 20, 2008]. Eur J Hum Genet. doi:10.1038/ejhg.2008.6.38. Wang F, Chang D, Hu FL, et al. DNA repair gene XPD polymorphisms and cancer risk: a meta-analysis based on 56 case-control studies. Cancer Epidemiol Biomarkers Prev. 2008;17(3):507-517.
FREE FULL TEXT
39. Figueroa JD, Malats N, Rothman N, et al. Evaluation of genetic variation in the double-strand break repair pathway and bladder cancer risk. Carcinogenesis. 2007;28(8):1788-1793.
FREE FULL TEXT
40. Lewis SJ, Smith GD. Alcohol, ALDH2, and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1967-1971.
FREE FULL TEXT
41. Yang CX, Matsuo K, Wang ZM, Tajima K. Phase I/II enzyme gene polymorphisms and esophageal cancer risk: a meta-analysis of the literature. World J Gastroenterol. 2005;11(17):2531-2538.
PUBMED
42. Wang GY, Lu CQ, Zhang RM, Hu XH, Luo ZW. The E-cadherin gene polymorphism 160C-A and cancer risk: A HuGE review and meta-analysis of 26 case-control studies. Am J Epidemiol. 2008;167(1):7-14.
FREE FULL TEXT
43. Boccia S, La Torre G, Gianfagna F, Mannocci A, Ricciardi G. Glutathione S-transferase T1 status and gastric cancer risk: a meta-analysis of the literature. Mutagenesis. 2006;21(2):115-123.
FREE FULL TEXT
44. Wang P, Xia HH, Zhang JY, Dai LP, Xu XQ, Wang KJ. Association of interleukin-1 gene polymorphisms with gastric cancer: ameta-analysis. Int J Cancer. 2007;120(3):552-562.
PUBMED
45. Boccia S, Hung R, Ricciardi G, et al. Meta- and pooled analyses of the methylenetetrahydrofolate reductase C677T and A1298C polymorphisms and gastric cancer risk: a huge-GSEC review. Am J Epidemiol. 2008;(5):505-516.46. Zhou Y, Li N, Zhuang W, et al. P53 codon 72 polymorphism and gastric cancer: a meta-analysis of the literature. Int J Cancer. 2007;121(7):1481-1486.
PUBMED
47. Gorouhi F, Islami F, Bahrami H, Kamangar F. Tumour-necrosis factor-A polymorphisms and gastric cancer risk: a meta-analysis [published online March 4, 2008]. Br J Cancer. doi:10.1038/sj.bjc.6604277.48. Tripathy CB, Roy N. Meta-analysis of glutathione S-transferase M1 genotype and risk toward head and neck cancer. Head Neck. 2006;28(3):217-224.
PUBMED
49. Ye Z, Song H, Guo Y. Glutathione S-transferase M1, T1 status and the risk of head and neck cancer: a meta-analysis. J Med Genet. 2004;41(5):360-365.
FREE FULL TEXT
50. Huang WY, Olshan AF, Schwartz SM, et al. Selected genetic polymorphisms inMGMT, XRCC1, XPD, and XRCC3 and risk of head and neck cancer: a pooled analysis. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1747-1753.
FREE FULL TEXT
51. Vineis P, Veglia F, Benhamou S, et al. CYP1A1 T3801 C polymorphism and lung cancer: a pooled analysis of 2451 cases and 3358 controls. Int J Cancer. 2003;104(5):650-657.
PUBMED
52. Shi X, Zhou S, Wang Z, Zhou Z, Wang Z. CYP1A1 and GSTM1 polymorphisms and lung cancer risk in Chinese populations: a meta-analysis. Lung Cancer. 2008;59(2):155-163.
PUBMED
53. Rostami-Hodjegan A, Lennard MS, Woods HF, Tucker GT. Meta-analysis of studies of the CYP2D6 polymorphism in relation to lung cancer and Parkinson’s disease. Pharmacogenetics. 1998;8(3):227-238.
PUBMED
54. Raimondi S, Paracchini V, Autrup H, et al. Metaand pooled analysis of GSTT1 and lung cancer: a HuGE-GSEC review. Am J Epidemiol. 2006;164(11):1027-1042.
FREE FULL TEXT
55. Wilkening S, Bermejo JL, Hemminki K. MDM2 SNP309 and cancer risk: a combined analysis. Carcinogenesis. 2007;28(11):2262-2267.
FREE FULL TEXT
56. Lee WJ, Brennan P, Boffetta P, et al. Microsomal epoxide hydrolase polymorphisms and lung cancer risk: a quantitative review. Biomarkers. 2002;7(3):230-241.
PUBMED
57. Taioli E, Benhamou S, Bouchardy C, et al. Myeloperoxidase G-463A polymorphism and lung cancer: a HuGE genetic susceptibility to environmental carcinogens pooled analysis. Genet Med. 2007;9(2):67-73.
PUBMED
58. Kiyohara C, Yoshimasu K. Genetic polymorphisms in the nucleotide excision repair pathway and lung cancer risk: ameta-analysis. Int JMed Sci. 2007;4(2):59-71.
59. Kiyohara C, Takayama K, Nakanishi Y. Association of genetic polymorphisms in the base excision repair pathway with lung cancer risk: a meta-analysis. Lung Cancer. 2006;54(3):267-283.
PUBMED
60. Han S, Zhang HT, Wang Z, et al. DNA repair gene XRCC3 polymorphisms and cancer risk: a metaanalysis of 48 case-control studies. Eur J Hum Genet. 2006;14(10):1136-1144.
PUBMED
61. Rothman N, Skibola CF, Wang SS, et al. Genetic variation in TNF and IL10 and risk of non-Hodgkin lymphoma: a report from the InterLymph Consortium. Lancet Oncol. 2006;7(1):27-38.
PUBMED
62. Gayther SA, Song H, Ramus SJ, et al. Tagging single nucleotide polymorphisms in cell cycle control genes and susceptibility to invasive epithelial ovarian cancer. Cancer Res. 2007;67(7):3027-3035.
FREE FULL TEXT
63. Zeegers MP, Kiemeney LA, Nieder AM, Ostrer H. How strong is the association between CAG and GGN repeat length polymorphisms in the androgen receptor gene and prostate cancer risk? Cancer Epidemiol Biomarkers Prev. 2004;13(11 pt 1):1765-1771.
FREE FULL TEXT
64. Li H, Tai BC. RNASEL gene polymorphisms and the risk of prostate cancer: ameta-analysis. Clin Cancer Res. 2006;12(19):5713-5719.
FREE FULL TEXT
65. Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a HuGE Review. Am J Epidemiol. 2005;162(10):925-942.
FREE FULL TEXT
66. Bethke L, Webb E, Murray A, et al. Comprehensive analysis of the role of DNA repair gene polymorphisms on risk of glioma. Hum Mol Genet. 2008;17(6):800-805.
FREE FULL TEXT
67. Brenner AV, Butler MA, Wang SS, et al. Singlenucleotide polymorphisms in selected cytokine genes and risk of adult glioma. Carcinogenesis. 2007;28(12):2543-2547.
FREE FULL TEXT
68. Ye Z, Song H. Glutathione s-transferase polymorphisms (GSTM1, GSTP1 and GSTT1) and the risk of acute leukaemia: a systematic review and meta-analysis. Eur J Cancer. 2005;41(7):980-989.
PUBMED
69. Pereira TV, Rudnicki M, Pereira AC. Pombo-de- Oliveira MS, Franco RF. 5,10-Methylenetetrahydrofolate reductase polymorphisms and acute lymphoblastic leukemia risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1956-1963.
FREE FULL TEXT
70. Bethke L, Murray A, Webb E, et al. Comprehensive analysis of DNA repair gene variants and risk of meningioma. J Natl Cancer Inst. 2008;100(4):270-276.
FREE FULL TEXT
71. Lee KM, Lan Q, Kricker A, et al. One-carbon metabolism gene polymorphisms and risk of non- Hodgkin lymphoma in Australia. Hum Genet. 2007;122(5):525-533.
PUBMED
72. Thomas DC, Clayton DG. Betting odds and genetic associations. J Natl Cancer Inst. 2004;96(6):421-423.
FREE FULL TEXT
73. Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30(6):445-600.
PUBMED
74. Yeh CC, Sung FC, Tang R, Chang-Chieh CR, Hsieh LL. Association between polymorphisms of biotransformation and DNA-repair genes and risk of colorectal cancer in Taiwan. J Biomed Sci. 2007;14(2):183-193.
PUBMED
75. Mart’nez C. Mart’n F, Ferna’ndez JM, et al. Glutathione S-transferases mu 1, theta 1, pi 1, alpha 1 and mu 3 genetic polymorphisms and the risk of colorectal and gastric cancers in humans. Pharmacogenomics. 2006;7(5):711-718.
PUBMED
76. Little J, Sharp L, Masson LF, et al. Colorectal cancer and genetic polymorphisms of CYP1A1, GSTM1 and GSTT1: a case-control study in the Grampian region of Scotland. Int J Cancer. 2006;119(9):2155-2164.
PUBMED
77. Rajagopal R, Deakin M, Fawole AS, et al. Glutathione S-transferase T1 polymorphisms are associated with outcome in colorectal cancer. Carcinogenesis. 2005;26(12):2157-2163.
FREE FULL TEXT
78. Ates NA, Tamer L, Ates C, et al. Glutathione Stransferase M1, T1, P1 genotypes and risk for development of colorectal cancer. Biochem Genet. 2005;43(3-4):149-163.
PUBMED
79. Bolufer P, Collado M, Barragan E, et al. The potential effect of gender in combination with common genetic polymorphisms of drug-metabolizing enzymes on the risk of developing acute leukemia. Haematologica. 2007;92(3):308-314.
FREE FULL TEXT
80. Vineis P, Veglia F, Garte S, et al. Genetic susceptibility according to three metabolic pathways in cancers of the lung and bladder and in myeloid leukemias in nonsmokers. Ann Oncol. 2007;18(7):1230-1242.
FREE FULL TEXT
81. Cengiz M, Ozaydin A, Ozkilic AC, Dedekarginoglu G. The investigation of GSTT1, GSTM1 and SOD polymorphism in bladder cancer patients. Int Urol Nephrol. 2007;39(4):1043-1048.
PUBMED
82. Bolufer P, Barragan E, Collado M, Cervera J, Lopez JA, Sanz MA. Influence of genetic polymorphisms on the risk of developing leukemia and on disease progression. Leuk Res. 2006;30(12):1471-1491.
PUBMED
83. Aydin-SayitogluM. Hatirnaz O, Erensoy N, Ozbek U. Role of CYP2D6, CYP1A1, CYP2E1, GSTT1, and GSTM1 genes in the susceptibility to acute leukemias. Am J Hematol. 2006;81(3):162-170.
PUBMED
84. Shao J, Gu M, Zhang Z, Xu Z, Hu Q, Qian L. Genetic variants of the cytochrome P450 and glutathione S-transferase associated with risk of bladder cancer in a south-eastern Chinese population. Int J Urol. 2008;15(3):216-221.
PUBMED
85. Majumdar S, Mondal BC, Ghosh M, et al. Association of cytochrome P450, glutathione Stransferase and N-acetyl transferase 2 gene polymor- phisms with incidence of acute myeloid leukemia. Eur J Cancer Prev. 2008;17(2):125-132.
PUBMED
86. Huang K, Sandler RS, Millikan RC, Schroeder JC, North KE, Hu J. GSTM1 and GSTT1 polymorphisms, cigarette smoking, and risk of colon cancer: a population- based case-control study in North Carolina (United States). Cancer Causes Control. 2006;17(4):385-394.
PUBMED
87. Clavel J, Bellec S, Rebouissou S, et al. Childhood leukaemia, polymorphisms of metabolism enzyme genes, and interactions with maternal tobacco, coffee and alcohol consumption during pregnancy. Eur J Cancer Prev. 2005;14(6):531-540.
PUBMED
88. Pakakasama S, Mukda E, Sasanakul W, et al. Polymorphisms of drug-metabolizing enzymes and risk of childhood acute lymphoblastic leukemia. Am J Hematol. 2005;79(3):202-205.
PUBMED
89. Nelson HH, Wiencke JK, Christiani DC, et al. Ethnic differences in the prevalence of the homozygous deleted genotype of glutathione S-transferase theta. Carcinogenesis. 1995;16(5):1243-1245.
FREE FULL TEXT
90. Rebbeck TR. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 1997;6(9):733-743.
FREE FULL TEXT
91. Hayes JD, Strange RC. Potential contribution of the glutathione S-transferase supergene family to resistance to oxidative stress. Free Radic Res. 1995;22(3):193-207.
PUBMED
92. Parl FF. Glutathione S-transferase genotypes and cancer risk. Cancer Lett. 2005;221(2):123-129.
PUBMED
93. Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506-507:65-77.
94. Hein DW, Doll MA, Fretland AJ, et al. Molecular genetics and epidemiology of the NAT1 and NAT acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9(1):29-42.
FREE FULL TEXT
95. Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10(1):111-113.
PUBMED
96. Rozen R.. Genetic predisposition to hyperhomocysteinemia: deficiency of methylenetetrahydrofolate reductase (MTHFR). Thromb Haemost. 1997;78(1):523-526.
PUBMED
97. Choi SW, Mason JB. Folate status: effects on pathways of colorectal carcinogenesis. J Nutr. 2002;132(8)(suppl):2413S-2418S.
FREE FULL TEXT
98. Ló pez-Cima MF, Gonza lez-Arriaga P, Garca-Castro L, et al. Polymorphisms in XPC, XPD, XRCC1, and XRCC3 DNA repair genes and lung cancer risk in a population of Northern Spain.BMCCancer. 2007;7:162.99. De Ruyck K, Szaumkessel M, De Rudder I, et al. Polymorphisms in base-excision repair and nucleotideexcision repair genes in relation to lung cancer risk. Mutat Res. 2007;631(2):101-110.
PUBMED
100. Pachouri SS, Sobti RC, Kaur P, Singh J. Contrasting impact of DNA repair gene XRCC1 polymorphisms Arg399Gln and Arg194Trp on the risk of lung cancer in the north-Indian population. DNA Cell Biol. 2007;26(3):186-191.
PUBMED
101. Yin J, Vogel U, Ma Y, Qi R, Sun Z, Wang H. The DNA repair gene XRCC1 and genetic susceptibility of lung cancer in a northeastern Chinese population. Lung Cancer. 2007;56(2):153-160.
PUBMED
102. Sreeja L, Syamala VS, Syamala V, et al. Prognostic importance of DNA repair gene polymorphisms of XRCC1 Arg399Gln and XPD Lys751Gln in lung cancer patients from India. J Cancer Res Clin Oncol. 2008;134(6):645-652.
PUBMED
103. Sun J, Turner A, Xu J, Gronberg H, Isaacs W. Genetic variability in inflammation pathways and prostate cancer risk. Urol Oncol. 2007;25(3):250-259.
PUBMED
104. Smith JR, Freije D, Carpten JD, et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996;274(5291):1371-1374.
FREE FULL TEXT
105. Shook SJ, Beuten J, Torkko KC, et al. Association of RNASEL variants with prostate cancer risk in Hispanic Caucasians and African Americans. Clin Cancer Res. 2007;13(19):5959-5964.
FREE FULL TEXT
106. Cybulski C, Wokolorczyk D, Jakubowska A, et al. DNA variation in MSR1, RNASEL and E-cadherin genes and prostate cancer in Poland. Urol Int. 2007;79(1):44-49.
PUBMED
107. Shea PR, Ishwad CS, Bunker CH, Patrick AL, Kuller LH, Ferrell RE. RNASEL and RNASEL-inhibitor variation and prostate cancer risk in Afro-Caribbeans. Prostate. 2008;68(4):354-359.
PUBMED
108. Momand J, Wu HH, Dasgupta G. MDM2– master regulator of the p53 tumor suppressor protein. Gene. 2000;242(1-2):15-29.
PUBMED
109. Liu G, Wheatley-Price P, Zhou W, et al. Genetic polymorphisms of MDM2, cumulative cigarette smoking and nonsmall cell lung cancer risk. Int J Cancer. 2008;122(4):915-918.
PUBMED
110. Glick AB. TGFbeta1, back to the future: revisiting its role as a transforming growth factor. Cancer Biol Ther. 2004;3(3):276-283.
PUBMED
111. Rosfjord EC, Dickson RB. Growth factors, apoptosis, and survival of mammary epithelial cells.JMammary Gland Biol Neoplasia. 1999;4(2):229-237.
112. Cox DG, Penney K, Guo Q, Hankinson SE, Hunter DJ. TGFB1 and TGFBR1 polymorphisms and breast cancer risk in the Nurses’ Health Study. BMC Cancer. 2007;7:175.
PUBMED
113. Grenet J, Teitz T, Wei T, Valentine V, Kidd VJ. Structure and chromosome localization of the human CASP8 gene. Gene. 1999;226(2):225-232.
PUBMED
114. Sigurdson AJ, Bhatti P, Doody MM, et al. Polymorphisms in apoptosis- and proliferation-related genes, ionizing radiation exposure, and risk of breast cancer among US Radiologic Technologists. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2000-2007.
FREE FULL TEXT
115. Gudmundsson J, Sulem P, Manolescu A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39(5):631-637.
PUBMED
116. Yeager M, Orr N, Hayes RB, et al. Genomewide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39(5):645-649.
PUBMED
117. Haiman CA, Patterson N, Freedman ML, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39(5):638-644.
PUBMED
118. Tomlinson I, Webb E, Carvajal-Carmona L, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39(8):984-988.
PUBMED
119. Haiman CA, Le Marchand L, Yamamato J, et al. A common genetic risk factor for colorectal and prostate cancer. Nat Genet. 2007;39(8):954-956.
PUBMED
120. Zanke BW, Greenwood CM, Rangrej J, et al. Genome- wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39(8):989-994.
PUBMED
121. Broderick P, Carvajal-Carmona L, Pittman AM, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39(11):1315-1317.
PUBMED
122. Easton DF, Pooley KA, Dunning AM, et al. Genome- wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148): 1087-1093.
PUBMED
123. Duggan D, Zheng SL, Knowlton M, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99(24):1836-1844.
FREE FULL TEXT
124. Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40(3):316-321.
PUBMED
125. Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40(3):310-315.
PUBMED
126. Gudmundsson J, Sulem P, Rafnar T, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40(3):281-283.
PUBMED
127. Hunter DJ, Kraft P, Jacobs KB, et al. A genomewide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39(7):870-874.
PUBMED
128. Stacey SN, Manolescu A, Sulem P, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39(7):865-869.
PUBMED
ARTICLE EN RAPPORT
Cette semaine dans le JAMA-Français
JAMA. 2008;299:2361.
Texte Complet
|