- Institution: Stanford Univ Med Ctr Lane Med Lib/Periodical Dept/Rm L109
- Sign In as Member / Individual
Neuropathic Pain: The Paradox of Dynorphin
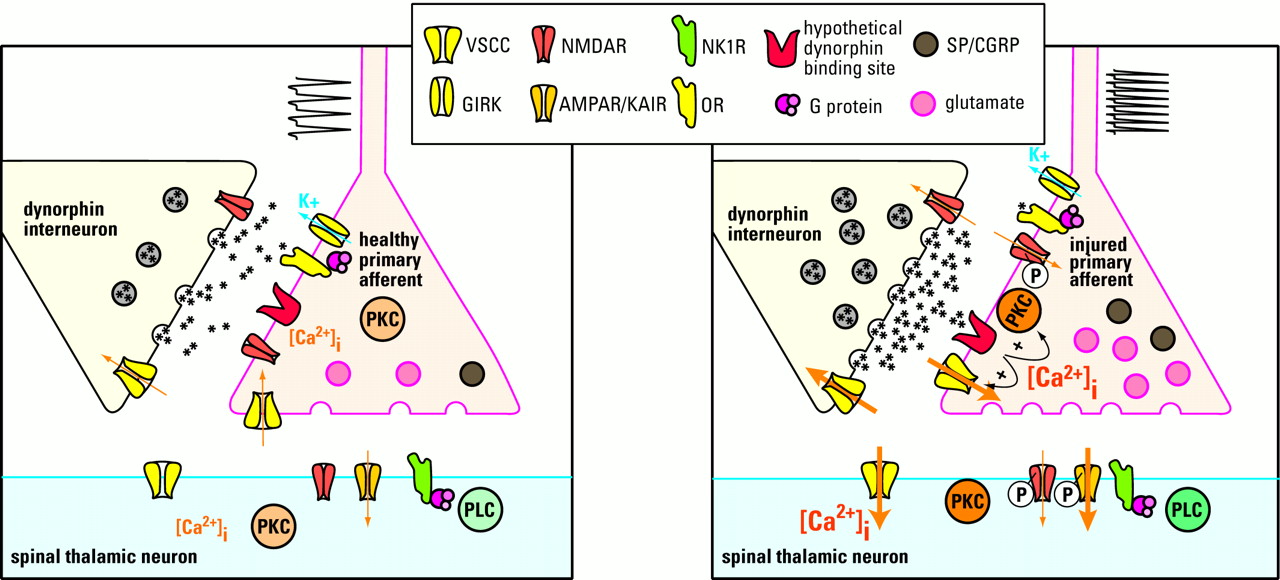
Possible mechanisms of dynorphin in maintaining neuropathic pain. Under normal conditions (left panel), dynorphin acts at opioid receptors (OR) to limit noxious input from a primary afferent: Specifically, dynorphin activates potassium efflux (through a G protein–coupled inwardly rectifying potassium channel; GIRK) and inhibits calcium influx (through a voltage-sensitive calcium channel; VSCC) in primary afferent and spinal thalamic neurons. Thus, excitatory neurotransmitter (glutamate) release is inhibited from the primary afferent terminal. After nerve injury (right panel), sustained input from the injured nerve drives the upregulation of spinal dynorphin through an unknown mechanism. The enhanced release of dynorphin stimulates the activity of molecules such as protein kinase C (PKC) and VSCC at the primary afferent to facilitate release of excitatory neurotransmitter and neuropeptides (e.g., substance P [SP] and CGRP). The binding site that mediates the effects of dynorphin is unknown. Enhanced neurotransmission potentiates the activity of postsynaptic glutamate receptors (AMPAR/KAIR) and NMDA receptors (NMDAR) by calcium/PKC dependent receptor phosphorylation. This is in part due to the activation of the substance P receptor (NK1R)/phospholipase C (PLC) pathway.

