A GABAB Mystery: The Search for Pharmacologically Distinct GABAB Receptors
Abstract
The classification of neurotransmitter receptors into distinct pharmacological subtypes is of major importance in drug discovery. This quest is particularly important for neurotransmitter systems that are widely distributed. Because γ-aminobutyric acid (GABA) receptors, both GABAA and GABAB, are found throughout the neuroaxis, they are likely involved in all central nervous system functions. Accordingly, the therapeutic promise of GABAB receptor manipulation depends upon the identification of subtypes than can be specifically targeted.

The preponderance of GABAergic activity in rat brain is revealed by in situ hybridization of mRNA that encodes the GABABR1 receptor. The development of drugs that can usefully modulate GABAB receptor activity hinges in part on efforts to identify and target pharmacologically distinguishable receptor subtypes.
[Image courtesy of Nature (www.nature.com) and B. Bettler. Nature 386, 239–246 (1997)]
INTRODUCTION
Target site selectivity is essential to the development of safe and effective therapeutics, and has been greatly facilitated by the cloning and characterization of neurotransmitter receptor subtypes. The identification of pharmacologically distinct receptor subtypes, such as the β1-adrenergic, 5-HT3, and histamine H1 and H2 receptors, has enabled the manipulation of specific organs and cells of therapeutic interest without adversely affecting other systems regulated by the same transmitter. Even within receptor subfamilies, it is possible to target certain receptors, as has been the case with 5-HT1A and 5HT1B/D agents. It has thus become virtually axiomatic that receptor systems are composed of molecularly and pharmacologically distinct subgroups.
GABA receptors, found in up to 40% of neurons in the central nervous system (1), can be broadly divided, on the basis of their molecular properties and effector systems, into two broad categories (2). Ionotropic, or GABAA, receptors are pentameric structures assembled from a family of distinct subunits (3). Activation of the GABAA site allows for the diffusion of chloride ion down its concentration gradient, typically resulting, in mature neurons, in an increased intracellular chloride concentration and hyperpolarization of the cell. With eighteen known subunits capable of combining to form GABAA receptors, hundreds of distinct pentameric subtypes could exist. In reality, only a limited number are found in the mammalian central nervous system, chiefly α1β2γ2. Subtyping of GABAA receptors has made it possible to develop drugs that selectively influence this transmitter system (4). Moreover, the possibility of identifying additional GABAA subunits and subunit combinations that mediate individual physiological responses increases the likelihood of designing even more selective agents (5).
The second major category of GABA receptors comprises those that are coupled to G proteins. Termed GABAB, stimulation of these receptors modifies adenylyl cyclase activity, decreases calcium conductance, and increases potassium conductance in neurons (Figure 1⇓) (6). Although their existence has been known for years (7), GABAB receptors have only recently been cloned (8–12), thereby renewing interest in their potential as therapeutic targets. Although the GABAB receptor has consequently revealed a number of highly unusual, if not unique, properties among G protein–coupled receptors, questions remain about whether it, like most other seven-transmembrane receptors, exists in molecularly and pharmacologically distinct forms. Details emerging from the quest to identify GABAB receptor subtypes are the focus of this review. Those wishing a more extensive discussion on other aspects of GABAB receptors are directed elsewhere (6, 13–15)
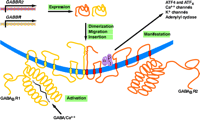
Schematic representation of GABAB receptor (GABABR) assembly and function. The biogenesis of functional GABABRs depends on distinct genes (GABBR1 and GABBR2), the expression of which results in distinct seven-transmembrane–domain products that undergo heterodimerization, followed by migration (trafficking) and insertion into the plasma membrane. Activation of the R1 protomer by GABA and Ca2+ is manifested through a number of transduction mechanisms that are mediated through association of the R2 protomer with a trimeric (α, β, γ) G protein.
THE PHARMACOLOGY OF GABAB RECEPTORS
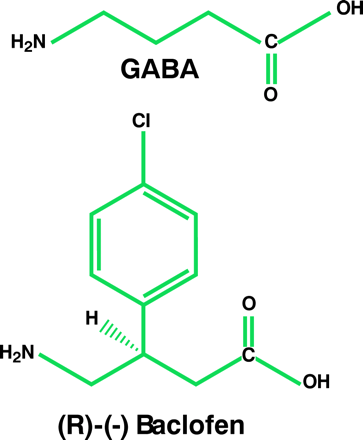
The GABAB receptor was first identified by studies aimed at defining the mechanism of action of baclofen (β-[4-chlorophenyl]-GABA), a muscle relaxant used to treat spasticity. These investigations revealed that baclofen—along with GABA—inhibits depolarization-induced neurotransmitter release from brain and peripheral tissues (7, 16), an effect that is not inhibited by bicuculline, a GABAA receptor antagonist. Soon after this discovery, it was demonstrated that baclofen influences adenylyl cyclase activity in a pertussis toxin–sensitive manner, suggesting that the drug stimulates a Gi/o protein–coupled receptor; GABAA activity, in contrast, is a ligand-gated ion channel (17–19). Electrophysiological studies revealed that baclofen causes a late inhibitory postsynaptic potential that is secondary to an increase in potassium conductance (20, 21). Moreover, activation of the GABAB receptor reduces excitatory postsynaptic potentials, presumably by modifying presynaptic calcium currents, which, in turn, decreases the release of excitatory neurotransmitters (20, 22). These actions further distinguished this receptor from the GABAA site.
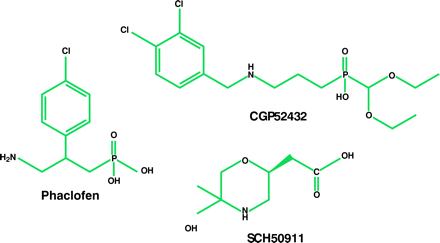
Characterization of the GABAB receptor was hindered initially by the paucity of potent and selective receptor agonists and antagonists. Eventually, a number of GABAB agonists were developed, including 3-aminopropyl-phosphinic acid (3-APPA) and CGP44532 (23, 24). Of major importance was the synthesis of selective receptor antagonists (25–27). The first of these, including phaclofen, saclofen, CGP35348, CGP36742, and SCH50911, did not manifest high affinity for GABAB receptors; however, their selectivity—as well as the ability of some to cross the blood–brain barrier—facilitated the characterization of this site. High-affinity GABAB receptor antagonists, such as CGP54626A, CGP55845A, and CGP52432, resulted from the addition of a 3,4-dichlorobenzyl or a 3-carboxybenzyl substituent onto the phosphinic acid function of the pharmacophore (26, 28). Moreover, the iodinated antagonists CGP64213 and CGP71872 were important tools for cloning GABAB receptor genes (29).
The development of agonists and antagonists has made possible a more precise assessment of the therapeutic utility of targeting the GABAB receptor. Besides the conventional use of baclofen to treat spasticity and skeletal muscle rigidity, GABAB agonists display antinociceptive activity in humans and in animal models of pain (30–34). Although baclofen is used clinically to treat neuropathic pain and, when administered intrathecally, to attenuate pain associated with spinal cord injury or stroke, its use as a general analgesic is limited because of its sedative properties and the rapid development of tolerance to its pain-relieving activity (35–37). On the other hand, laboratory and clinical studies indicate that GABAB agonists reduce craving for a number of addictive substances, including cocaine and opioids (38–48). Baclofen also displays antitussive activity, anti-ischemic activity on heart muscle, and inhibits intestinal motility and vagally mediated bronchoconstriction (41–44).
There are as yet no clinical data on the pharmacological responses to GABAB receptor antagonists, although laboratory studies imply these agents may possess anticonvulsant, antidepressant, and antipsychotic activities (45–47). In addition, GABAB receptor antagonists enhance cognition and stimulate the production of growth factors in brain, suggesting they may have neuroprotective properties (48–51). The range of responses elicited by antagonists and agonists have further underscored the importance of identifying molecularly distinct GABAB receptors so as to fully exploit their pharmacological potential.
CHARACTERIZATION OF GABAB RECEPTORS
Pharmacological Evidence of Subtypes
A variety of methods have been used in the search for pharmacologically distinct GABAB receptors. Although a distinction between high- and low-affinity GABAB receptor binding in rat brain was taken as an indication of pharmacologically distinct sites (52, 53), heterogeneous binding characteristics may simply reflect different conformational states of a single receptor protein. Thus, studies involving GABAB receptor–mediated responses provide somewhat more direct evidence of receptor subtypes. For example, the affinities of GABAB receptor antagonists have been reported to differ with respect to their ability to block baclofen-induced inhihition of GABA, glutamate, cholecystokinin, and somatostatin-like immunoreactivity release from cerebrocortical synaptosomes (54–56); however, these findings have been open to dispute (57).
Receptor subtyping has also been attempted by investigating the effect of GABAB agonists and antagonists on second messenger responses in rat brain slices (58, 59). In these studies, advantage was taken of the fact that baclofen can either inhibit or enhance cAMP production, depending upon the pre-existing state of the adenylyl cyclase system (60). Specifically, whereas baclofen inhibits forskolin-stimulated cAMP production, it augments cAMP synthesis in the presence of substances, such as isoproterenol, that liberate Gαs. Thus, it has been proposed that the inhibitory effect of baclofen reflects the coupling of a GABAB receptor subtype with Gi, and that the augmenting effect arises from a GABAB receptor subtype that is coupled to Go (6, 61). Although the inhibition-to-stimulation ratio with regard to adenylyl cyclase activity proved to be constant among a group of GABAB agonists, some antagonists inhibited the effect of baclofen on the isoproteronol-mediated process significantly more potently than on its effect on the forskolin-mediated response (59).
Intracellular recordings have revealed that pre- and postsynaptic GABAB receptors differ with respect to their sensitivity to N-ethylmaleimide, and electrophysiological experiments suggest that pre- and postsynaptic GABAB receptors respond differently to certain agonists and antagonists (62–64). Together, these biochemical and physiological studies indicate the existence of GABAB receptor subtypes. Indeed, some of these data point to the possibility that presynaptic GABAB receptors, which regulate neurotransmitter release, differ from the postsynaptic sites responsible for the late inhibitory potential. Nonetheless, because these results provide only indirect evidence for receptor subtypes, definitive conclusions await the structural identification of molecularly distinct GABAB receptors.
Cloning and Structural Identification
Early attempts to clone the gene that encodes the GABAB receptor were unsuccessful. Ultimately, a novel high-affinity radiolabeled antagonist was used to identify protein expressed from a rat cDNA library (65). Curiously, although the overexpressed receptor bound appreciably to GABAB receptor antagonists, it bound relatively poorly to agonists. In addition, only a small fraction of the recombinant protein that was expressed could be located on plasma membranes in transfected cells (65), leading to the speculation that the GABAB receptor required a trafficking protein for delivery to the cell surface. Subsequent work revealed the existence of a second protein, whose sequence, moreover, was related (54% amino acid similarity and 35% sequence homology) to that of the cloned GABAB receptor itself (9–12). Most striking was the discovery that the second protein is required not only to establish full expression of GABAB receptor activity, including appropriate sensitivity to agonists, but that it does so by forming a stable heterodimeric association with the cloned receptor that persists, beyond mere “trafficking,” within the cell membrane (Figure 1⇑). The originally cloned GABAB receptor molecule was subsequently termed GABAB1, and the second, related protein has been termed GABAB2. Although each protein subunit displays some GABAB receptor activity when present alone, it appears, at least in transfected cell expression systems, that only the heterodimer is a fully operative and appropriately sensitive to GABAB agonists (66–68).
Although it had been known for some time that G protein–coupled receptors are capable of forming homodimers (69–71), not until the discovery of the GABAB heterodimer was it possible to demonstrate directly that dimerized seven-transmembrane receptors can be essential for receptor function. It has subsequently been demonstrated that opioid receptors also form functional heterodimers (72). Because early work on GABAB receptor heterodimers utilized only recombinant systems, questions remained about whether GABAB1–GABAB2 dimerization is required for wild-type receptors. Studies utilizing a clonal line of mouse melanotropes (mIL-tsA58), rat pituitary intermediate lobe melanotropes, and rat dorsal root and superior cervical ganglion neurons demonstrated that both subunits are absolutely required in native receptors as well (73–76).
It appears that heterodimerization of the two GABAB receptor proteins occurs predominantly through association of the alpha-helical portions of the two C termini, and that this association is essential for trafficking of the receptor (Figure 1⇑) (77). It further appears that the large N-terminal extracellular domain—in particular, that of the GABAB1 subunit—is the site for ligand binding. The GABAB2 subunit, on the other hand, is crucial for effector coupling (Figure 1⇑) (78–80). Modeling and mutation studies suggest a “Venus flytrap” model for GABAB receptor activation, with agonist causing closure of the binding domain (81). Structurally, the GABAB receptor is closely related to vomeronasal organ receptors, metabotropic glutamate receptors, and calcium-sensing receptors (82). The similarity with calcium-sensing sites is particularly intriguing because calcium ion is required for the binding of agonist to the GABAB receptor (Figure 1⇑) (79, 83).
THE GABAB HETERODIMER: DOES IT TAKE TWO TO TANGO?
The initial cloning studies from rat brain revealed two isoforms of the GABAB1 subunit: GABAB1a and GABAB1b (8). Subsequently, four additional splice variant isoforms (c-f) of this subunit, and three of the GABAB2 subunit, were identified (Table 1⇓) (84). Not all of these isoforms are capable of forming functional GABAB receptors when linked to GABAB2. The GABAB1e isoform, for example, is thought to bind to, and thus limit, the availability of GABAB2, thereby regulating the number of functional GABAB receptors (84).
Rat GABAB Receptor Subunits
The absolute functional requirement for GABAB1–GABAB2 heterodimerization is demonstrated by studies with GABAB1-knockout mice, which are devoid of GABAB receptor activity (45, 85). Such data might suggest that GABAB1 and GABAB2 would be regulated and expressed in unison to maintain the normal complement of functional receptor. Accordingly, it is perplexing that this does not appear to be the case. Specifically, in response to a peripheral nociceptive stimulus, levels of GABAB1 and GABAB2 mRNA in the rat dorsal lumbar spinal cord rise differentially (86). In the rat dorsal root ganglia, a pain stimulus increases GABAB1 mRNA levels, whereas GABAB2 mRNA content remains low. Furthermore, levels of GABAB2 mRNA in rat spinal cord decline significantly from birth to 21 days of age, during which time GABAB1 mRNA levels remain constant (87). The observed, differential regulation of the genes encoding the two subunits is difficult to reconcile with the proposition that GABAB receptors are exclusively composed of GABAB1 and GABAB2 subunits. It must thus be considered that formation of GABAB receptors may be only one function—perhaps even a minor function—of these proteins.
The differential expression of GABAB1 and GABAB2 is also reflected in their distribution in brain and other tissues. Whereas in situ hybridization studies suggest there are brain regions containing much greater amounts of GABAB1 than GABAB2 mRNA, the relative levels of these proteins appears fairly constant throughout the brain (9, 88–90). The relatively constant ratio of GABAB1 to GABAB2 protein in the presence of variable mRNA ratios suggests brain regional differences in message or protein stability. Likewise, although GABAB1a and GABAB1b proteins are detectable in the rat anterior pituitary, very little GABAB2 is present (91). Moreover, despite the obvious existence of GABAB1a and GABAB1b message in many peripheral organs, such as heart, spleen, lung, liver, intestine, stomach, and urinary bladder, GABAB2 message is less detectable in these tissues (92, 84). Conversely, GABAB2 message, but not GABAB1 message, is found in the retina (67). Inasmuch as GABAB1 requires GABAB2 to migrate to the cell membrane, at least in recombinant systems, the mechanism for transporting GABAB1 in peripheral tissues remains unknown.
Both GABAB1a and GABAB1b mRNA is localized to neuronal cells, but not glial cells, in brain (93). Both mRNAs are found throughout the brain, although their preponderance, relative to one another, varies among brain regions. For example, the level of GABAB1a mRNA is two- to threefold greater than that of GABAB1b mRNA in the pars reticulata of the substantia nigra and in the mesencephalic reticular nucleus, whereas GABAB1b mRNA is threefold and tenfold more prominent than GABAB1a mRNA in the medial septum and Purkinje cells, respectively (93). In terms of subcellular localization, presynaptic GABAB receptors appear to be predominantly GABAB1a–GABAB2, whereas GABAB1b–GABAB2 dimers seem to be located mainly at postsynaptic sites (88, 93–95). Because the GABAB1a protein is also found postsynaptically, however, it is not possible to generalize about functional distinctions between the isoforms (96). Although GABAB receptor subunits and GABAB receptor binding are found throughout the central nervous system, the receptor isoform composition varies with the brain region. Indeed, it has been suggested that differences in isoform combinations may yield pharmacologically distinct sites.
DIFFERENTIAL INTERACTIONS BETWEEN GABAPENTIN AND GABAB ISOFORMS
The discovery that the heterodimeric GABAB receptor arises from the expression of only two genes would seem to limit the possibility of pharmacologically distinct receptor subtypes. Furthermore, the fact that the GABA binding site sequence is the same for all GABAB1 isoforms argues against the existence of pharmacologically distinct subtypes. To test the hypothesis that the dimeric combination of alternative splice variants could result in altered pharmacological selectivities toward agonists and antagonists, recombinant receptors consisting of either the GABAB1a–GABAB2 or the GABAB1b–GABAB2 association have been examined. In general, the results have failed to support any significant pharmacological differences between these isoform combinations (11, 97). An exception occurs with regard to gabapentin, an anticonvulsant that appears to be a selective agonist for the GABAB1a–GABAB2 heterodimer (98, 99). This conclusion is based on studies with recombinant GABAB receptors overexpressed in oocytes, as well as wild-type receptors in mIL-tsA58 cells and rat hippocampal brain slices. In all cases, the response of the GABAB1a–GABAB2 dimer to gabapentin is blocked by a GABAB receptor antagonist. These results underscore the possibility of developing therapeutic agents that might pharmacologically differentiate between GABAB receptor isoform combinations.
Although these in vitro data suggest that gabapentin is a selective agonist for the GABAB1a–GABAB2 dimer, in vivo studies fail to confirm this notion, at least with respect to some of its pharmacological effects. Thus, gabapentin induces neuronal inhibition (i.e., paired-pulse inhibition) independent of GABAB receptors and, unlike baclofen and the GABAB agonist CGP35024, the analgesic response to gabapentin in rat is not inhibited by a GABAB receptor antagonist (100, 101). These results suggest that gabapentin is not solely a GABAB agonist; some effects, such as its antinociceptive action are mediated by an interaction with other sites, such as the α2δ subunit of voltage-dependent calcium channels (102). Nevertheless, the anticonvulsant or mood stabilizing effects of gabapentin might result from a selective interaction with a GABAB receptor subtype.
THE SEARCH CONTINUES
It has been over two decades since the GABAB receptor was first identified, and three years since it was cloned. Dozens of GABAB receptor agonists and antagonists have been synthesized, and the effector systems for this neurotransmitter receptor have been identified and characterized. Given these advances, it is surprising the question of pharmacologically distinct subtypes remains unresolved. A reason for this persisting uncertainty may be that the vast majority of GABAB receptor agonists and antagonists are of the same chemical class, so that they would be of little use in differentiating among receptor subclasses. Compounds that display some selectivity for GABAB receptor subclasses, such as gabapentin and γ-hydroxybutyrate, manifest relatively low affinity and lack of sufficient selectivity, making it difficult for them to provide conclusive results with respect to GABAB receptor subtype selectivity (98, 103). As long as the data suggesting pharmacologically distinct GABAB receptor are derived solely from in vitro experiments, it may not be possible to establish a pharmacologically and clinically meaningful distinction of receptor subtypes. Clearly, a greater variety of systemically active GABAB receptor agonists and antagonists would help resolve this issue. Admittedly, the design of chemical agents capable of distinguishing between the GABAB1a–GABAB2 and GABAB1b–GABAB2 sites may be difficult, given that the GABAB1 isoforms differ only in areas not thought to be associated with ligand binding.
The possibility remains that GABAB receptor subtypes result from an association of either GABAB subunit with other, structurally unrelated proteins, which could, in turn, affect pharmacological specificity. Evidence for this is provided by the finding that GABAB1 and GABAB2 expression is regulated independently of one another, and that the distribution of these proteins differs among brain areas and peripheral tissues (86, 88–90). In addition, immunoprecipitation studies suggest that other, as yet unidentified, proteins may associate with the GABAB receptor (88). Because GABAB1 knockout studies indicate this subunit is absolutely essential for a functioning GABAB receptor, it appears that subtypes are most likely to be composed of this subunit and a protein that can substitute, both as a chaperone and in coupling to the appropriate effector systems, for GABAB2 (45, 85).
Yeast two-hybrid studies have demonstrated that both the GABAB1 and GABAB2 subunits associate with other proteins, in particular transcription factors CREB2 (ATF4) and ATFx (Figure 1⇑) (104). Furthermore, GABAB receptor stimulation leads to nuclear accumulation of CREB2 and resulting transcriptional activation in neuronal cell cultures, suggesting that these subunits may be involved in regulating long-term changes in the central nervous system (104, 105).
Although knockdown experiments using a mouse pituitary cell line suggest that both GABAB1 and GABAB2 are absolutely required for GABAB receptor function (80), it seems possible that other proteins capable of dimerizing with GABAB subunits to form a functional receptor are not constitutively expressed. Thus, the induction of ancillary, regulatory proteins in response to certain conditions must be considered. In any event, the search for proteins that can regulate or serve as GABAB receptor subunits is integral to the quest for pharmacologically distinct GABAB receptor activities. Peripheral tissue known to respond to baclofen and to lack GABAB2 subunits might be a particularly rich source of such entities.
CONCLUSIONS
Research on GABA receptors has yielded new insights into neurotransmitter receptor systems in general. Studies of GABAA receptors reveal how drugs, such as benzodiazepines, allosterically regulate ligand-gated ion channels in therapeutically meaningful ways. Likewise, GABAB receptor research has provided new information on G protein–coupled receptors and receptor crosstalk (6). Moreover, GABAB receptors were the first wild-type seven-transmembrane heterodimers to be identified, and the GABAB heterodimers remain one of the few major receptor systems for which pharmacologically distinct subtypes have not been firmly established. Drug development in this area requires that efforts continue to identify the manner in which such subtypes might be formed.
- © American Society for Pharmacology and Experimental Theraputics 2001
References

S.J. Enna, Ph.D., is Chair of the Department of Pharmacology, Toxicology and Therapeutics at the Kansas University School of Medicine. He is Editor of the Journal of Pharmacology and Experimental Therapeutics, and is Past President of ASPET. E-mail senna{at}kumc.edu; fax 913-588-7501.

