DNA DAMAGE-INDUCED MUTAGENESIS
A NOVEL TARGET FOR CANCER PREVENTION
Abstract
Tolerance to some degree of unrepaired DNA damage is crucial for cell survival—more specifically, for the sustained functionality of the DNA replication machinery—in the presence of adverse (genotoxic) conditions. At least two mechanisms ensure such tolerance: template switching and lesion bypass. Lesion bypass, whereby unrepaired damaged DNA serves as template, involves the Y family of DNA polymerases; lesion bypass can be error-free or error-prone, depending on the nucleotide incorporated during translesion synthesis. Error-prone lesion bypass constitutes a major mechanism of mutagenesis and, in eukaryotes, is primarily effected by the DNA polymerase ζ (Polζ) pathway. A relationship between the Y family polymerases and the Polζ pathway is thus implicated, and conforms to the two-polymerase two-step model of lesion bypass. Based on the mutagenesis hypothesis of cancer formation, DNA damage–induced mutagenesis and its underlying molecular biology offer an intriguing potential target for cancer prevention.
INTRODUCTION
DNA is frequently damaged, both by endogenous and environmental agents. In general, four complex systems have evolved to respond to DNA damage: 1) DNA repair; 2) cell cycle checkpoint control; 3) apoptosis; and 4) damage tolerance. By removing lesions from DNA, DNA repair forms the most effective defense system against DNA damage and comprises at least five mechanisms: a) base excision repair (BER); b) nucleotide excision repair (NER); c) mismatch repair; d) recombinational repair; and e) direct reversal of damage. Endogenous (or “spontaneous”) DNA lesions are mainly repaired by BER, whereas NER is an important mechanism for removing a wide spectrum of damage, especially bulky DNA lesions that cause significant structural distortions. Mismatch repair corrects mismatched bases, small deletions, and small insertions that result from errors of replicative DNA polymerases. Recombinational repair is required to repair double-stranded DNA breaks and is thus especially important in response to ionizing radiation. Direct reversal of damage is a highly specialized repair mechanism. In humans, only the MGMT (O6-methylguanine-DNA methyltransferase) protein is known to function by this repair mechanism, and irreversibly accepts (i.e., noncatalytically), the methyl group directly from O6-methylguanine from within DNA. A list of human DNA repair genes was recently compiled by Wood et al. (1).
In response to DNA damage, the progression of the cell cycle into S phase is delayed by the G1 cell cycle checkpoint control, whereas progression into M phase is halted by the G2 checkpoint. Prolongation of the G1 and G2 phases functions to permit more effective DNA repair and thus avoids DNA synthesis and mitosis in the presence of excessive DNA damage. Like DNA repair, cell cycle checkpoint control promotes genomic stability and cell survival following DNA damage (2). In contrast, apoptosis in response to DNA damage is a mechanism that eliminates cells with heavily damaged DNA, thus protecting the genomic integrity of multicellular organisms. For example, sunburn caused by skin cell apoptosis is regarded as a protective mechanism against UV-induced neoplastic transformation (3).
Even when DNA repair and cell cycle checkpoint control are fully functional, some DNA lesions often persist through replication of the genome. Factors that contribute to the persistence of DNA damage include: a) high levels of damage; b) poorly repaired lesions; c) inefficiently repaired genomic regions; and d) DNA damage incurred during the S phase of the cell cycle. Because many lesions that persist despite DNA repair and cell cycle checkpoints hamper or thwart the replication apparatus, cells have evolved a damage tolerance system to allow complete replication in the presence of DNA damage. This response tolerates, rather than removes, DNA damage, and consists of at least two mechanisms: a) template switching and b) lesion bypass. Lesion bypass greatly increases the likelihood of mutations. In fact, error-prone lesion bypass constitutes the major mechanism of DNA damage–induced mutagenesis in cells. In this review, our current understanding of damage tolerance is summarized with emphasis on lesion bypass mechanisms, and the concept of targeting damage-induced mutagenesis for cancer prevention is presented.
DAMAGE TOLERANCE
Damage tolerance is a measure of last resort to rescue cells from DNA damage. Without the damage tolerance response, cells would become highly sensitive to killing by DNA-damaging agents, as is exemplified by yeast rad18 (radiation sensitive) mutant cells (4). In the yeast S. cerevisiae, where the damage tolerance response is best understood among all eukaryotes, Rad6 and Rad18 are required for both mechanisms of damage tolerance, that is, template switching and lesion bypass (5, 6). Rad6 is a ubiquitin-conjugating enzyme (7) and forms a complex with Rad18 (8). The Rad6–Rad18 complex is thought to function at an early step in both mechanisms of the damage tolerance response (6, 8). Humans contain two Rad6 homologs, HHR6A and HHR6B (9). Recently, the human RAD18 gene has been identified and cloned (10, 11). The human RAD18 protein also interacts and forms a complex with HHR6A or HHR6B (10,11). Furthermore, expression of a mutant human rad18 cDNA in cultured human cells leads to cellular sensitivity to several DNA damaging agents, presumably as a result of a dominant negative effect of the mutant RAD18 protein (11). Thus, it appears that damage tolerance systems similar to those of yeast also function in humans.
Template Switching
Template switching has also been referred to as “postreplication repair” (12); however, because the damage is tolerated rather than repaired, the term “postreplication repair” will be avoided in this review. Template switching in mammalian cells was originally proposed by Higgins et al. (13) to describe the case where, although normal synthesis of dsDNA is blocked by a lesion on one of the template strands, synthesis on the undamaged template strand may nevertheless continue to a limited extent. Then, by using the newly synthesized daughter strand as template (template switching), the replicative machinery effectively circumvents the lesion-blocked DNA and proceeds with replication. Following dissociation of the two newly synthesized daughter strands, each is re-annealed to its original parental strand to effect semiconservative replication. Having passed the lesion site, the replication apparatus can then resume normal DNA synthesis (Figure 1⇓). Template switching avoids replication of the damaged site of the DNA template; therefore, the newly synthesized daughter DNA strand is error-free opposite the lesion.
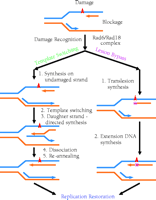
Two mechanisms of damage tolerance. Template switching is error-free, because it avoids copying the damaged site of the DNA template. Lesion bypass directly uses damaged DNA as template. Consequently, mutations (shown as a purple squares) are often generated opposite the lesion site.
In addition to the Rad6–Rad18 complex, Rad5, Mms2, (methyl methanesulfonate sensitive) Ubc13 (ubiquitin conjugating), PCNA (proliferating cell nuclear antigen), and DNA polymerase δ (Polδ) are involved in template switching in yeast (5, 14–,18). Recently, physical interactions have been demonstrated between the following protein pairs: Mms2–Ubc13 (16), Ubc13–Rad5 (19), and Rad5–Rad18 (19). Thus, Rad5 may serve to recruit the Mms2–Ubc13 complex to the Rad6–Rad18 complex at site of DNA damage (19). (See Figure 2⇓.) Whereas Mms2 is a Ubc-like protein (15), Rad5 is a ring finger protein containing conserved helicase motifs (14). Rad5 does in fact possess a DNA-dependent ATPase activity, but a DNA helicase activity has not been detected (20). The molecular mechanism of template switching remains largely undefined.
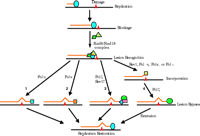
A lesion bypass model. The large oval represents replication proteins. Numbers 1–4 indicate four different lesion bypass mechanisms. The two-polymerase two-step hypothesis for lesion bypass is depicted as mechanism 4, and the Pol ζ dual-function hypothesis is illustrated in mechanisms 3. Squares in mechanisms 1 and 2, Polη and Polκ, respectively; green oval in mechanisms 3 and 4, Polζ; rectangle and diamond in mechanism 3, putative accessory factors of Polζ.
Lesion Bypass
Unlike template switching, lesion bypass directly utilizes the damaged template. Conceptually, lesion bypass can be divided into two steps: i) nucleotide incorporation opposite the lesion (i.e., translesion synthesis), followed by ii) extension of DNA synthesis. After a short stretch of extension, normal DNA synthesis by the replication apparatus can then resume (Figure 1⇑). Significantly, lesion bypass can be either error-free, whereby the correct nucleotide opposite the damage is predominantly incorporated, or error-prone, whereby an incorrect nucleotide is frequently incorporated opposite the damage. Consequently, error-free lesion bypass is a mutation-avoiding mechanism, and error-prone lesion bypass is a mutation-generating mechanism.
Whereas the Rad6 and Rad18 proteins are involved in both template switching and lesion bypass, it is completely unknown how cells choose one mechanism vs. the other. Nevertheless, in view of the differing probabilities for mutagenesis, the commitment that the cell makes to one or the other mechanism has profound biological consequences. It is clear that the two parallel mechanisms provide cells with a functional redundancy for tolerance to DNA damage during replication, thereby enhancing the ability of cells to survive unrepaired genomic lesions.
Lesion bypass requires a specialized DNA polymerase (Pol) that can use damaged DNA as template. Earlier genetic studies of damage-induced mutagenesis in yeast identified several pertinent genes, one of which was REV3 (required for reversion mutation) (21). In 1989, Morrison et al. (22) recognized that the REV3 gene product is probably a DNA polymerase. Seven years later, Nelson et al. (23) experimentally confirmed that the Rev3 protein is indeed a DNA polymerase, which they named Polζ, and established the connection between Polζ and lesion bypass. Very recently, a new family of DNA polymerases, known as the Y family (also known as the UmuC superfamily [Umu for Unmutable]), has emerged (24, 25). Biochemical studies suggest that the whole Y family of DNA polymerases is probably involved in translesion synthesis (25).
THE Y FAMILY OF DNA POLYMERASES
The prototypic members of the Y family include E. coli DNA polymerase IV (pol IV), E. coli pol V, Rev1 of yeast, Polη of yeast, and human Polι (24, 25). E. coli pol IV is encoded by the dinB (damage inducible) gene (26). Initially, pol IV was found to play an important role in “untargeted mutagenesis” in E. coli (27). “Untargeted mutagenesis” refers to induced mutations that arise in undamaged regions of DNA. More recent genetic analyses suggest that pol IV is also involved in both error-free and –1 frameshift translesion syntheses of benzo[a]pyrene-damaged DNA (28). E. coli pol V is encoded by the umuC gene (29, 30). Genetic and biochemical studies have unequivocally established that pol V is the translesion synthesis polymerase of the SOS mutagenesis pathway, the major error-prone lesion bypass mechanism in E. coli (29–,33). During lesion bypass, pol V functions in the form of the UmuD'2-UmuC protein complex (29, 30). The umuC and umuD genes (encoding pol V) and the dinB gene (encoding pol IV) are tightly controlled by the SOS response system, which is switched on by DNA-damaging agents (33). Therefore, lesion bypass activity in E. coli is controlled at the level of gene expression. Furthermore, pol V is additionally controlled posttranslationally through the cleavage of the non-functional UmuD to the smaller functional UmuD' (33).
DNA Polymerase η
Polη is encoded by the RAD30 gene in yeast and the XPV (POLH) gene in humans (34–,36), and plays an important role in response to UV radiation. Yeast cells lacking Polη exhibit increased sensitivity to UV radiation (37, 38), whereas in humans, defects of the Polη-encoding gene will lead to the hereditary disease xeroderma pigmentosum variant (XPV) (35, 36). XPV patients exhibit sensitivity to sunlight and a predisposition to skin cancer (39). Unlike other XP patients (i.e., XPA through XPG), who are deficient in NER, XPV patients are proficient in NER but deficient in DNA replication following UV radiation (40), and XPV cells are thus hypermutable by UV (41–,43), which explains the predisposition for skin cancer in XPV patients.
In vitro, purified Polη is able to efficiently bypass thymine-thymine dimers (TT dimers) in an error-free manner (34, 35, 44), and it is the loss of this biochemical activity that defines the molecular pathology of the XPV disease (35, 36). Initially, Polη was believed to be an error-free translesion synthesis polymerase specific to UV radiation. More recently, Yuan et al. (45) have demonstrated that purified yeast Polη is capable of error-free nucleotide incorporation opposite a template guanine containing an acetylaminofluorene (AAF) bulky adduct. This result was later confirmed with purified human Polη (46). Consistent with these biochemical results, XPV cell-free extracts are deficient in the translesion synthesis of AAF adducts (47). Human Polη can also effectively catalyze error-prone synthesis in vitro opposite template (+)-trans-anti-benzo[a]pyrene-N2-dG adducts, which are bulky template lesions (48). The (+)-trans-anti-benzo[a]pyrene-N2-dG bulky adduct is the major lesion formed in cells after exposure to the potent carcinogen benzo[a]pyrene, and is highly mutagenic in COS cells (49, 50). Human Polη predominantly incorporates an A, less frequently a T, and least frequently a G or a C at the sequence context examined (47). This specificity of nucleotide incorporation correlates well with the mutagenic specificity of this lesion in COS cells (49, 50), thereby supporting a role for Polη in error-prone bypass of the (+)-trans-anti-benzo[a]pyrene-N2-dG lesion in mammalian cells.
DNA Polymerase ι
Human Polι is very unique in that it violates the Watson-Crick base-pairing rule opposite the T of the template strand(56–,58). This polymerase incorporates G opposite T with three- to tenfold higher rates than it does A (56–,58), although the resulting T–G base pair results in a poorer substrate for Polι relative to normal Watson-Crick–paired substrate. Consequently, DNA elongation by human Polι frequently aborts opposite the template T, a property that is designated as the “T stop” (56); human Polι also has a low catalytic efficiency opposite template C (56, 57). As a result of sluggish catalysis opposite template pyrimidines, human Polι is able to synthesize only very short stretches of DNA. These unique features suggest that Polι may play a specialized function in cells, such as somatic hypermutation during immunoglobulin development (56, 58). Somatic hypermutation of rearranged heavy and light chain variable regions of antibody-coding genes is characterized by enormous mutation rates on the order of 10-3–10-4 per base pair per generation, about 106-fold higher than that in other genes in mammalian cells (59). The hypermutation polymerase responsible for generating this extreme mutation rate otherwise remains elusive.
Several biochemical studies indicate that Polι is capable of both error-free and error-prone translesion syntheses in vitro. In response to 8-oxoguanine (a major product of oxidative damage) in DNA, human Polι predominantly incorporates C opposite the lesion, and is also able to incorporate the correct C opposite the AAF-adduct of guanine; however, further DNA synthesis is blocked by these lesions (60). Interestingly, human Polι–mediated incorporation of nucleotides is more efficient at template AP sites than it is opposite template T (56), and is characterized by the nucleotide specificity: G>T>A>C (57, 60). Reminiscent of the T stop, DNA synthesis is aborted following the incorporation of a single nucleotide opposite AP sites (57, 60).
Surprisingly, human Polι preferentially incorporates A opposite the 3' T of template TT (6-4) photoproducts (57,60, 61), in contrast to its usual (non-Watson-Crick) preference for incorporating G opposite undamaged template T (56–,58). Nucleotide incorporation opposite the 5' T of the TT (6–4) photoproduct, however, is largely prohibited (56, 57). Opposite template cyclobutane TT dimers, human Polι has a very limited activity, preferentially incorporating a T opposite the 3' T of the lesion (60, 61). This activity, albeit very inefficient, may contribute to TT dimer–induced mutagenesis in XPV cells that lack Polη (61), in light of the fact that the 3' T of the TT dimer completely blocks purified yeast Polζ (57, 62).
A Polι homolog has been found in Drosophila (63), but not in yeast. A truncated Drosophila Polι, missing 288 C-terminal amino acids, incorporates A more readily than it does G opposite undamaged template T, preferentially incorporates A opposite the 3' T of the TT (6-4) photoproduct (albeit with a low efficiency), and catalyzes efficient error-free bypass of TT dimers (63). It is not clear whether the C-terminal truncation significantly affects the biochemical properties of Polι, or whether the Drosophila Polι manifests a substrate specificity that is distinct from human Polι.
DNA POLYMERASE κ
Human Polκ efficiently bypasses template (-)-trans-anti-BPDE-N2-dG adducts (BPDE for benzo[a]pyrene diol epoxide) in an error-free manner by incorporating C opposite the lesion (64). Because (-)-trans-anti-BPDE-N2-dG is a bulky DNA adduct formed by cellular exposure to benzo[a]pyrene, Polκ may play an important role in suppressing benzo[a]pyrene-induced mutagenesis in humans (64). Human Polκ efficiently bypasses 8-oxoguanine in an error-prone manner, preferentially incorporating A opposite the lesion (64). Opposite a template AP site, purified human Polκ most frequently incorporates A (64, 65). Efficient extension from this Polκ-incorporated A requires T as the next template base (i.e., 5' to the AP site), and is mediated mainly by a –1 deletion mechanism (64, 65), thereby suggesting that Polκ re-aligns the terminal A incorporated into the nascent strand (i.e., across from the AP site) with the next template T (64, 65). In support of this model, the placement of an A, C, or G 5' to the AP site of the template greatly reduces Polκ translesion activity (64, 65). Furthermore, without a template T 5' to the AP site, lesion bypass, albeit inefficient, occurs mainly by the Polκ-mediated insertion of A followed by extension without template realignment (64, 65). Opposite an AAF-modified guanine, human Polκ slightly favors incorporation of T over incorporation of C (64–,66). Less frequently, A is incorporated opposite the lesion, and G incorporation occurs least frequently (64, 65). This specificity of nucleotide incorporation is, in general, consistent with the in vivo mutagenesis results of Shibutani et al. (67), who transformed COS cells with a shuttle vector containing a site-specific AAF-guanine and sequenced the replicated plasmid clones. Thus, Polκ may contribute to AAF-induced mutagenesis in mammalian cells. Error-free AAF bypass by Polη may provide an explanation for the fact that the majority of recovered shuttle vector from COS cells did not contain mutations opposite the lesion (67). These studies indicate that Polκ, although not involved in response to UV damage, likely functions otherwise as a translesion polymerase, and can do so in an error-free or error-prone manner, depending on the specific lesion.
The Rev1 dCMP Transferase
In 1996, Nelson et al. made the milestone discovery that the yeast Rev1 protein is a DNA template–dependent deoxycytidyl (dCMP) transferase (23). Three years later, attempts to experimentally confirm the expectation that Rad30 (a homolog of Rev1) might possess a similar activity established, in fact, that the yeast Rad30 protein is DNA Polη (34). Discoveries of yeast Polη, E. coli pol IV, and E. coli pol V in 1999 (26, 29, 30, 34) led the way to the explosive studies on the Y family of DNA polymerases in 2000.
Yeast Rev1 efficiently incorporates dCMP opposite G and AP sites (23), and is thus probably responsible for the in vivo bypass of AP sites in yeast cells that results predominantly in C incorporation (68, 69). Rev1 is unable to respond to template TT dimers (69); its activity opposite other DNA lesions is not known.
The human REV1 gene has been identified and cloned (70, 71). Lin et al. demonstrated that the dCMP transferase activity is conserved in the human REV1 protein in that it efficiently incorporates dCMP opposite template G, U, and AP sites (70). The enzyme's ability to recognize template G, U, and AP sites, despite their structural disparities, is intriguing, especially in view of its strict specificity for dCMP insertion. On a template of polydG, human REV1 is able to perform nucleotide incorporation and extension, synthesizing a daughter strand of polydC (Y. Zhang and Z. Wang, unpublished results). Thus, REV1 may be considered as a specialized DNA polymerase.
All in the Family
In addition to the sequence similarity among the members of the Y family of DNA polymerases, a common functionality has become apparent: They are all capable of translesion synthesis, at least in vitro. For a given DNA lesion, translesion synthesis can be error-free or error-prone, depending on the polymerase involved; conversely, for a given translesion polymerase, nucleotide incorporation can be error-free or error-prone, depending on the specific lesion (Table 1⇓).
Translesion specificity of human Polη, Polι, and Polκ
It appears that each polymerase of the Y family is associated with a unique DNA lesion specificity, although some lesions, such as AP sites, may be recognized by multiple polymerases. Such functional redundancy likely provides better efficiency of translesion synthesis during DNA replication. More important, the unique range of template utilization manifested by each polymerase underscores the importance of evolving multiple DNA polymerases for translesion synthesis. The notion of unique functionalities predicts that the loss of even one translesion polymerase could be sufficient to trigger health problems in humans. Indeed, XPV disease, in which Polη activity is lost, demonstrates that members of the Y family can be crucial (35, 36). In vitro biochemical studies have been vital in characterizing translesion synthesis by the Y family of DNA polymerases. However, in vivo genetic analyses in the near future will be indispensable to definitively establish that all of these polymerases are indeed recruited for translesion synthesis in cells.
THE POLζ MUTAGENESIS PATHWAY
The Polζ mutagenesis pathway is a major error-prone lesion bypass mechanism in yeast. Proteins required in the Polζ mutagenesis pathway include Rad6, Rad18, Rev1, Rev3, and Rev7 (4, 22, 72–,75). Most recently, Huang et al. (76) found that the Pol32 subunit of yeast Polδ, a replication polymerase, is also involved in the Polζ mutagenesis pathway. Most likely, more genes are involved in this mutagenesis pathway, such as the REV6 gene identified sixteen years ago by genetic screening (77).
The Rev3 protein is the catalytic subunit of DNA Polζ (78). Rev7 strongly interacts with Rev3 to stimulate its polymerase activity, and is thus considered to be a proper subunit of Polζ (78). Since the cloning of the yeast REV3 gene in 1989 (22), it had been widely believed that Rev3 was probably the only polymerase responsible for certain instances of translesion synthesis. This notion was firmly held until 1999, when Polη was discovered and subsequently precipitated studies, not only of Polη, but also of Polι and Polκ. Surprisingly, purified yeast Polζ can be very inefficient in performing translesion synthesis in vitro (57, 62). Yeast Polζ is able to perform limited lesion bypass of a template TT (6-4) photoproduct, incorporating A or T, and less frequently G, opposite the 3' T; A is predominantly paired with the 5' T of the lesion (62). Limited translesion synthesis by yeast Polζ is also observed opposite AAF-modified guanine, with predominant incorporation of G (62). In E. coli, lesion bypass by pol V is stimulated by RecA, SSB, and the β subunit (processivity clamp) and the γ subunit (clamp loader) of polymerase III holoenzyme, (30–,31). Thus, the possibility that other factors may stimulate lesion bypass by Polζ must be ascertained.
The human REV3 and REV7 genes have been cloned (79–,81). The REV3 gene product is predicted to be a DNA polymerase (79, 80) and interacts with the human REV7 protein (81). Reduced expression of REV1 and REV3 genes by anti-sense RNA in cultured human cells results in significant reduction of UV-induced mutagenesis (71, 79). Ubiquitous expression of RAD18, REV1, and REV3 genes in various human tissues is consistent with the notion that the Polζ pathway may represent a major mutagenesis (error-prone lesion bypass) mechanism in mammals, including humans (10, 70, 80).
Beyond its essentiality to translesion synthesis opposite AP sites (23, 69), Rev1 may play additional roles in lesion bypass, although these have been only partially investigated. Because UV-induced mutagenesis also requires Rev1 (69, 72, 82), it was proposed that Rev1 might play anothr role, presumably noncatalytic, in the Polζ mutagenesis pathway, independent of its dCMP transferase activity (69, 82). Human REV1 can interact with Rev7; Rev7 homodimers have also been observed (83). The functional significance of these physical interactions is not yet clear. However, physical interactions identified among RAD6, RAD18, REV1, REV3, and REV7 suggest that lesion bypass by the Polζ mutagenesis pathway likely involves protein complexes at sites of DNA damage.
THE TWO-POLYMERASE TWO-STEP THEORY OF LESION BYPASS
Is there a relationship between the Y family of translesion polymerases and the Polζ mutagenesis pathway in lesion bypass? Clearly, although Polη, Polι, and Polκ can insert nucleotides opposite several DNA lesions in vitro, they are often very inefficient in the extension phase of DNA synthesis. The replication of damaged genomes would thus necessitate additional DNA polymerase activity(ies) that could extend synthesis past the lesion site. In yeast Rev1 studies, Nelson et al. (23) demonstrated that yeast Rev1 and Polζ together can achieve AP site bypass in vitro. More recently, Yuan et al. (45) have shown that AP sites can be sequentially bypassed in vitro by nucleotide insertion opposite the lesion by yeast Polη followed by DNA extension by yeast Polζ. These crucial observations led to the proposal that some lesion bypass in eukaryotes may involve the concerted actions of one polymerase (e.g., Polη) for translesion nucleotide insertion, and subsequent extension of the nascent strand by Polζ (45). This theory is now referred to as the “two-polymerase two-step” model of lesion bypass (62). Further supporting this lesion bypass model, human Polι and yeast Polζ together could bypass a template AP site and TT (6-4) photoproduct (57).
Most likely, Polζ plays dual functions during lesion bypass in cells: nucleotide incorporation opposite some lesions; and extension of nascent DNA following translesion nucleotide insertion by other polymerases (62). The two-polymerase two-step action would thus functionally connect the Y family of polymerases to the Polζ pathway for purposes of lesion bypass, which further underscores the importance of the Polζ pathway in damage-induced mutagenesis in eukaryotes.
Although the bypass of many lesions may involve the two-polymerase two-step mechanism, some lesions could be bypassed efficiently by a single DNA polymerase; for example, Polη might be effective in bypassing TT dimers, and Polκ could similarly negotiate (-)-trans-anti-BPDE-N2-dG adducts (Figure 2⇑). Indeed, in response to UV radiation, Polη is genetically (epistatically) related to the Rad6–Rad18 function, but is largely unrelated to the Rad5 and the Polζ functions (37). It is thus conceivable that multiple mechanisms exist in cells to bypass various lesions (Figure 2⇑). The presence of multiple translesion polymerases raises an important question: How is each polymerase appropriately recruited to the site(s) of DNA damage where it is (often uniquely) capable of accomplishing translesion synthesis? The answer to this question will be crucial to a better understanding of DNA lesion bypass.
LOW-FIDELITY CHARACTER OF TRANSLESION POLYMERASES
The DNA replicative fidelity of E. coli DNA Pol V is much lower than is that of Pol III on undamaged template (30, 31). In eukaryotes, Polη, Polι, and Polκ are all associated with extraordinarily low fidelity in copying undamaged DNA templates (56–58, 84–,88). The low-fidelity character of these polymerases is believed to be an inevitable consequence of their biological function in translesion synthesis. As proposed by the loose-and-flexible active site model (64, 85, 89), lesion bypass polymerases may have evolved to contain a loose and flexible active site that can accommodate a variety of lesions so that substrate specificity with regard to (damaged) template is broadened. Thus, undamaged template would fit the active site “pocket” loosely—without the stringent geometry constraints that characterize highly accurate Watson-Crick base pairing—and thereby result in extraordinarily low-fidelity DNA synthesis.
Apparently, these specialized low-fidelity polymerases are excluded from normal DNA replication in order to maintain genomic stability. Although the precise mechanism for their exclusion in this regard is not known, four mechanisms may be postulated. First, expression of these polymerases may be maintained at a low level under normal growth conditions. Indeed, the E. coli DNA polymerases IV and V are controlled by the SOS response system (33), and expression of Polη in yeast is induced upon cellular exposure to DNA damaging agents (37, 38). Second, the translesion synthesis function of these polymerases may be further controlled by a posttranslational mechanism. In E. coli, cleavage of the UmuD protein is regarded as an important mechanism controlling the activity of pol V in lesion bypass (33). Amazingly, a fundamentally similar mechanism also appears to be employed by eukaryotes: A nuclear localization sequence is located in the C-terminal region of Polη. Deleting this region does not affect its polymerase activity, but renders Polη biologically inactive in response to UV radiation (90, 91). Indeed, recombinant Rev1, Polη, Polι, and Polκ are all unstable, readily undergoing proteolysis at their C-terminal regions (Y. Zhang and Z. Wang, unpublished results). This C-terminal proteolysis may be an important mechanism for controlling the function of these lesion bypass polymerases. Third, access of the polymerase to the sites of DNA lesions during replication may be limited by a recruitment mechanism. Fourth, DNA syntheses by these polymerases are generally distributive (i.e., only one or a few nucleotides are synthesized before the polymerase dissociates from the template), thereby limiting their activity to the site(s) of lesion.
FUNDAMENTAL BASIS OF CANCER: THE MUTAGENESIS HYPOTHESIS
Cancer is an extremely complex disease, developing as a multi-stage process involving many genetic alterations. Cancer cells often exhibit abnormalities in cell proliferation, differentiation, and genomic stability. Genomic instability of cancer cells can include limited DNA sequence alterations (i.e., micro-instability) such as point mutations, deletions and insertions, or gross chromosomal alterations (i.e., macro-instability) such as chromosomal rearrangements and aneuploidy. Transformation of tissue-specific normal cells into the lethal metastatic cells involves drastic phenotypic changes.
What are the molecular bases for cellular transformation in cancer development? Extensive studies of cancer biology have identified a long list of genes whose malfunction contributes to cellular abnormalities in genomic stability, proliferation, differentiation, and mobility. Cellular functions affected by these genes include replication, recombination, DNA repair, transcription, signal transduction, cell cycle checkpoints, and apoptosis. The complexity of the molecular pathology of cancer is unmatched by any other human disease. It has been proposed that a mutator (increased mutation rate) phenotype may be required for tumor progression (92). A similar concept is presented here as the “mutagenesis hypothesis” for cancerous transformation, which postulates that the common foundation of most, if not all, cancer is mutagenesis (Figure 3⇓).
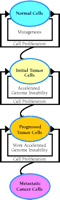
The mutagenesis hypothesis of cancer formation. This hypothesis postulates that the foundation of most cancer is mutagenesis. Mutations accumulate as cells proliferate. When mutations alter critical genes, tumorigenesis is inititated, likely involving genomic instability. Disruption of the normal mechanisms guarding genomic stability would in turn accelerate genome destabilization, leading to tumor progression and metastasis. Genomic instability includes gene mutations and chromosomal aberrations.
Cell proliferation and DNA damage are of great importance to mutagenesis. Without cell proliferation, DNA damage cannot be fixed into mutations and the limited fidelity of replicative polymerases cannot be “expressed.” During DNA replication, replicative DNA polymerases copy some three billion base pairs of human genome with impressive, but nevertheless limited, fidelity (93). Although the majority of polymerase errors are corrected by mismatch repair, the repair system itself does not function with 100% efficiency. Especially when the microsatellite regions of the genome are copied, polymerase errors are much higher due to template slippage (93), and some errors are likely to escape correction by the mismatch repair system. Furthermore, some unrepaired DNA damage, spontaneously formed or induced by environmental agents, will be responded to by error-prone lesion bypass, consequently leading to mutations. Hence, it is certain that mutations are generated each time the cell replicates itself. Agents that promote cell proliferation per se may not cause DNA damage directly, but indirectly cause mutagenesis by driving quiescent cell into replication; thus, such agents represent risk factors for cancerous transformation. DNA damage formed spontaneously or induced by environmental agents constitutes a major source of mutagenesis.
Initial mutations (the cancer foundation) that alter the normal function of genes involved in cell proliferation, repair, apoptosis, replication, signal transduction, cell cycle checkpoints, etc., including proto-oncogenes and tumor suppressor genes, will likely initiate carcinogenesis (tumor initiation). Cancer cells are often associated with chromosomal abnormalities that likely result from mutations in critical genes involved in maintaining chromosome stability. For example, mutations in RFC5 or MEC1 (i.e., two genes involved in DNA metabolism–related checkpoints) in yeast drastically increase chromosomal aberrations (94). The initial mutation of critical genes creates a momentum of further genomic instability, including chromosome aberrations, that can culminate in tumor progression and lead to alterations of additional genes critical for metastasis (Figure 3⇑). The accumulation of genetic alterations during cancer development would thus be an accelerating, rather than a linear, process. With the mutagenesis hypothesis in mind, DNA damage-induced mutagenesis offers a novel molecular target for cancer prevention.
INHIBITING DAMAGE-INDUCED MUTAGENESIS FOR CANCER PREVENTION
According to the mutagenesis hypothesis of carcinogenesis, mutation is a prerequisite for most, if not all, cancers. Thus, a novel strategy for cancer prevention would be to inhibit damage-induced mutagenesis (82, 95). If mutagenesis can be inhibited with a therapeutic agent, human cancer risk may be significantly reduced (Figure 4⇓). An attractive molecular target for inhibiting damage-induced mutagenesis is the Polζ mutagenesis pathway.
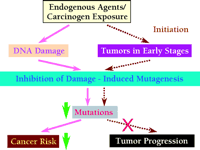
A novel strategy for cancer prevention. If the pathway(s) that underlie damage-induced mutagenesis can be inhibited by a therapeutic agent, significant decreases in cancer risk can be expected (solid line). Decreasing mutation rates could also block or slow down tumor progression, achieving cancer suppression (dashed line).
In yeast, the Polζ mutagenesis pathway is a major mechanism for both induced and spontaneous mutagenesis (Figure 2⇑) whereas the contribution it makes to cell survival in the presence of DNA damage appears to be small, probably due to efficient error-free lesion bypass and template switching in the absence of the Polζ pathway (22, 82). Thus, DNA damage should not become prohibitively cytotoxic upon inhibition of the Polζ mutagenesis pathway. Because the Polζ mechanism of mutagenesis functions as a concerted pathway, the inactivation of one component of the pathway should effectively prevent the formation of the end product (i.e., damage-induced mutations). Thus, REV1, REV3, and REV7 genes or their gene products are potential targets for inhibiting damage-induced mutagenesis. Of course, multiple targets would facilitate the successful development of effective agents to inhibit mutagenesis.
Using yeast as a model system, Rajpal et al. (82) were able to mimic the effects of Polζ inhibition by reducing cellular REV3 expression through molecular techniques. Indeed, lower levels of Polζ significantly reduced UV-induced mutation frequency. Using anti-sense RNA techniques, Gibbs et al. (71, 79) also showed that reducing the expression of REV1 or REV3 in cultured human cells can reduce UV-induced mutagenesis. These studies provide strong support for the concept of targeting Polζ and REV1 in order to inhibit DNA damage–induced mutagenesis with the ultimate goal of preventing neoplastic transformation and cancer.
CONCLUSIONS
Fighting cancer by hitting its very foundation, mutagenesis, is a novel concept in anti-cancer drug research. Exciting progress has been made in recent years in our understanding of DNA lesion bypass and damage-induced mutagenesis. Yeast has served very well as a eukaryotic model system in the studies of damage-induced mutagenesis. It is remarkable that our current understanding of the human Polζ pathway proteins and activities are directly and exclusively derived from yeast studies that began about thirty years ago (21). Much more insight into the mechanisms of damage-induced mutagenesis will undoubtedly come from future studies of the yeast model system, and indeed much more needs to be learned. Although, knocking out Polζ in mice results in embryonic lethality (96–,98), transgenic mice carrying mutations in the genes that underlie the Polζ pathway and encode the Y family of polymerases will also be important tools for modeling damage-induced mutagenesis in humans. Targeting DNA damage-induced mutagenesis for cancer prevention is likely a highly challenging task. However, the potential rewards of this anti-cancer strategy are enormous.
- © American Society for Pharmacology and Experimental Theraputics 2001
References

Zhigang Wang, Ph.D., is an Assistant Professor of the Graduate Center for Toxicology at the University of Kentucky, where he has received a Burroughs Wellcome Fund New Investigator Award to study mechanisms of DNA damage–induced mutagenesis.E-mail zwang{at}pop.uky.edu; fax 859-323-1059.

