Pharmacologically Directed Cell Disposal: Labeling Damaged Cells for Phagocytosis as a Strategy against Acute Pancreatitis
Apoptosis is the main mechanism of programmed cell deletion in multicellular organisms and proceeds with typical changes in cell morphology that accompany cell death and clearance. Necrosis, on the other hand, is a form of traumatic cell death featuring cell swelling, disruption of plasma membrane and the membranes of the organelles, and release of cytosolic constituents (Box 1). In contrast to apoptosis, which often provides beneficial effects to the organism, necrosis is almost always detrimental. Acute pancreatitis (AP) is a localized inflammation in the pancreas that occasionally evolves into a systemic life-threatening disease. Gallstones and alcohol consumption are the main risk factors of the development of AP. At present, there is no clinically effective pharmacological treatment for AP. During AP, acinar cells can die through both apoptosis and necrosis; however, the severity of AP correlates with the extent of necrotic cell death in pancreatic acini, and interventions promoting apoptosis rather than necrosis are hypothesized to be protective against the disease (1). Thus, in the context of AP, apoptosis and necrosis are competing death pathways. In multicellular organisms, removal of apoptotic cells by specialized phagocytes or bystander cells is normally accompanied by the release of anti-inflammatory mediators that prevent unwanted immune reaction to cytoplasm constituents derived from the dying cells (2). By contrast, cell necrosis triggers pro-inflammatory signaling cascades. During AP, local inflammatory reactions, if strong, progress into a systemic inflammatory response syndrome that is eventually responsible for the majority of AP morbidity and mortality.
Apoptosis, Autophagy, and Necrosis
Apoptosis is the main mechanism of programmed cell deletion that involves a series of biochemical events leading to a characteristic cell morphology (including chromatin condensation, loss of membrane asymmetry, blebbing, and cell shrinkage), death and total removal. Autophagy is a catabolic process of sequestration and degradation of a cell’s own components which helps the starving cell redirect the nutrients from dispensable to more-essential processes. Autophagy resulting in the total destruction of the cell is called type 2 programmed cell death. Necrosis is a form of traumatic cell death featuring cell swelling, disruption of plasma membrane and the membranes of the organelles and release of cytosolic constituents. In contrast to apoptosis, which often provides beneficial effects to the organism, necrosis is almost always pathological.
Although apoptosis is a perfect example of the programmed demise of a cell, it is not clear to what extent cell necrosis during AP is the result of programming rather than sudden, unregulated death caused by an overwhelming stress. Necrosis and apoptosis share some important intracellular signaling mechanisms, including activation of death receptors and recruitment of the associated adaptor proteins (such as FADD), increased concentrations of intra-cellular calcium, mitochondrial depolarization, and cytochrome c release, as well as activation of cathepsins and calpains (3, 4). These events take place in a well-regulated manner during apoptosis and might also be controlled during necrosis. During AP, the decreased expression or activity of caspases can result in a paradoxical shift from apoptosis to necrosis rather than from apoptosis to cell rescue (5). In many cell types, death with autophagic phenotype or by apoptosis develops in a mutually exclusive manner, so that inhibition of apoptosis facilitates autophagic death while inhibition of autophagy may induce apoptosis (6). During AP, autophagy is induced very early in pancreatic acinar cells (7) and appears to be an important mechanism of intracellular trypsin activation (8). Indirect evidence suggests that a particular type of programmed cell death featuring necrotic morphology called necroptosis, which depends on death domain receptor-associated adaptor kinase RIP-1 (9) and RIP-3 (10), may be implicated in AP (5). Another type of programmed necrosis relies on mitochondrial proteins cyclophilin D and BNIP3 and competes with RIP-dependent necrosis and may also participate during AP, as the molecular components of this necrotic pathway are found in the acinar cells (11).
Thus, the necrotic death of pancreatic acinar cells during AP may result from a controlled process, which can be slower, equally fast, or faster than that of the apoptotic program. Implicit in this suggestion is that a “slow” apoptosis may be overtaken by a “fast” necrosis (12). In addition, unsuccessful clearing of apoptotic cells by phagocytes may result in cell death by secondary necrosis. These scenarios illustrate the difficulty of finding a treatment that, by acting on the apoptotic cascade, would yield the right balance between cell survival and apoptosis to the detriment of necrosis. On the one hand, a manipulation that favors apoptosis may also add to cell stress and push the cell into necrosis. On the other hand, decreasing the likelihood of apoptosis may unexpectedly make necrosis kinetically favorable. Thus, some of the prospective drugs tested in animal models of AP or in clinical settings have been targeted against necrosis-specific mechanisms (e.g., PARP inhibitors) or autophagy (e.g., chloroquine), whereas others have been aimed generally to improve the cells’ well-being by reducing the cell stress (e.g. with ROS scavengers, PLA2 inhibitors) or by inhibiting inflammatory signaling (e.g., specific for p38 MAP kinase inhibitors; antibodies or soluble receptors specific for pro-inflammatory cytokines; PAF inhibitors; or anti-inflammatory cytokines) (13).
The facilitation of dead-cell removal (in particular, that of necrotic cells) might be an alternative approach for reducing the amount of necrosis, also resulting in active suppression of pancreatic inflammation during AP. The idea is to label necrotic cells with specific apoptotic “eat-me” markers so that the dying cells can be recycled in a manner specifically seen with apotosis.
Apoptotic cells are quickly recognized, internalized and degraded by specialized phagocytes and, more generally, by many types of epithelial cells, which can engulf and destroy encountered apoptotic bodies as well. Successful uptake of an apoptotic cell requires recognition of “eat-me” markers exposed at the surface of that cell by specific receptors at the phagocyte’s membrane, resulting in the formation of the “engulfment synapse.” The two most well-studied “eat-me” signals are phosphatidylserine (PS), a phospholipid found not only on the inside of the plasma membrane of normal cells but also on the outer leaflet of the plasma membrane of dying cells, and calreticulin (CRT), primarily an ER resident protein whose complex with another ER resident protein, ERp57, is often displayed at the cell surface during apoptosis (14, 15). Other molecules that may participate in dead-cell recognition include phosphatidyle-thanolamine calnexin (which is often exposed along with CRT), annexin I, HSP-70, HSP-90, and some carbohydrates [see (16) for review]. “Don’t eat-me” signals also exist, which include CD-31 and CD-47 (16).
CRT and PS serve somewhat different and mutually complementary roles. The role of PS in recognition of apoptotic cells is complicated by its presence at the outer membrane of some healthy cells, where this presence does not trigger an attack of the immune cells (17). On some occasions, PS exposure may take place and be recognized by macrophages also in cells dying via necrosis (18, 19) or autophagy (20). CRT is found on the surface of healthy living cells more often than is PS (21). During apoptosis it concentrates into membrane patches also enriched with PS, where these molecules provide cooperative stimuli for host engulfment (21). CRT exposure precedes that of PS in anthracycline- or ionizing irradiation–induced apoptosis; however, apoptosis arising from ER stress seems not to provoke CRT exposure [(22), however, see (15)]. Although in most cases the engulfment of apoptotic cells is accompanied by the release of anti-inflammatory mediators or proceeds immunologically silently, anthracycline- or ionizing irradiation–induced apoptosis of tumor cells can result in an immune response. CRT is necessary but not sufficient for immunogenicity of those apoptotic cells (22), and CRT favors uptake of apoptotic cells by dendritic cells rather than macrophages (23). Similar to CRT, HSP70 also has high immunogenicity (24). Recognition of “eat me” ligands is executed by endocytic receptors on phagocytes, including: CD36/αvβ3 complex; putative PS receptor molecules TIM-1 and TIM-4; receptor tyrosine kinase Mer; β2-glycoprotein-I receptor; complement receptors 3 and 4; and the members of the scavenger receptor family (class A macrophage scavenger receptor SR-A1, lectin-like oxidized low-density lipoprotein particles receptor-1, CD68, and stabilin-2) (16, 25). Recognition of these transmembrane signal molecules is further facilitated by bridging molecules called opsonins.
The molecular mechanisms of clearing necrotic cells are less known. They do not compete with those that clear apoptotic cells, although several macrophage receptor systems are used for recognition of both apoptotic and necrotic cells (26, 27). Soluble factors released by necrotic cells (such as HMGB1, SAP130, inflammatory cytokines, etc.) are recognized by Toll-like receptors, C-type lectin domain family 4 member E (CLEC4E, also termed Mincle), receptor of advanced glycation end-products (RAGE), and CLEC9A [also termed (DNGR-1 DC, NK lectin group receptor-1)] on the surface of phagocytes (28–30). Sodium urate crystals resulting from release of intracellular uric acid can directly interact with the cell membrane and, thus, activate dendritic cells (31). Additionally, exposure of α2,6-sialylated lactosaminic chains on the cell surface universally marks a cell for primary necrotic death, irrespective of the cell type (32). However, the distribution of α2,6-sialylated lactosaminic chains in apoptotic cells is predominantly cytosolic. The clearance of necrotic cells occurs through macropinocytosis as opposed to “zipper”-like engulfment of the apoptotic cells (33).
The rate of clearance of apoptotic cells varies substantially even for cells of the same type depending on the exact nature of apoptotic stimulus (34). In some instances, the efficiency of clearance may be so great that the apoptotic phenotype has little time to develop, whereas the efficiency in other cases is so meager that secondary necrosis develops. Insufficient clearance of apoptotic cells and the secondary necrosis have been found in patients with autoimmune diseases (35, 36). Several lines of evidence suggest that in AP the uptake of the dead cells is compromised.
In AP, as in some other settings (37), the clearance of dying cells occurs in a highly proteolytic environment. The proteolytic cleavage of the putative “eat-me” molecules or their binding receptors by cathepsins G, thrombin, and trypsin (37) or by elastase (38) significantly inhibits the uptake of dying cells by phagocytic cells. The main target of the proteases is presumably the phosphatidylserine receptor (PSR) on the surface of PMNs (37, 38); however, the proteolysis of some other molecules cannot be completely excluded. Therefore, it is feasible that during AP, the efficiency of the phagocytosis of dying pancreatic acinar cells is compromised, owing to the spillage of the pancreatic zymogens.
Lipoprotein oxidation during AP (39) delays the engulfment but not the binding of apoptotic cells by macrophages (40). AP is associated with increased expression of protein tyrosine phos-phatase SHP-1 (41), whose activation inhibits Fc (receptors for immunoglobulins) mediated phagocytosis (42) and phagocytosis of apoptotic cells (21). Crambene induces pancreatic acinar cell apoptosis and this induction was suggested to be therapeutically protective against AP (1); however, the clearance of these apoptotic cells by macrophages is extremely slow and secondary necrosis is often observed (43, 44).
The main residing phagocytes in the pancreas are pancreatic stellate cells (45), although the acinar cells, which normally do not express the PSR (46), also show some phagocytic activity towards apoptotic bodies (47). Some cytokines produced during AP, including tumor necrosis factor α and IL-1, suppress the phagocytic activity of individual pancreatic stellate cells (45). This effect can be reversed by treatment with the antidiabetic drug troglita-zone, a ligand of the peroxisome proliferator–activated receptor γ (PPARγ). Moreover, the engulfment of necrotic acinar cells by the stellate cells results in necrosis of these stellate cells themselves, dramatically reducing the number of living pancreatic stellate cells at certain time frame (48). Thus, similar to that of infiltrating macrophages, the phagocytic activity of resident pancreatic stellate cells is likely to be substantially inhibited during AP. Accordingly, the speed of removal of apoptotic (and probably necrotic) cells in pancreas is likely slow enough to give rise to additional inflammation, so that speeding up the clearing process could have beneficial effects.
Labeling cells with “eat-me” signals and enhancing their interaction with their receptors could be an effective way of boosting removal of any specifically targeted cells (21, 49). Therefore, inducing the expression of markers for phagocytosis has been suggested as a promising strategy of attacking tumors (49), although it has yet to be applied in practice (50, 51). This approach can be extended by changing the delivery technique (from stimulation of endogenous expression to administration of exogenous “eat-me” molecules fitted with cell-specific tags). Another natural extension for this approach is to apply it to dying cells (preferably necrotic) in acute inflammatory diseases such as AP. Addition of protease or lipase inhibitors may prove to be helpful to accelerate further the clearance of necrotic cells.
For this strategy, prospective drug molecules could consist of two blocks: a marker for phagocytosis and a specific pancreatic acinar cell–binding fragment, ideally recognizing the stressed cells (Figure 1). As mentioned before, PS (or less lipophilic phospho-L-serine) and CRT may serve as complementary “eat-me” labels. Therefore, if conjugated to different ligands only partly specific for pancreatic acinar cells, their use in combination might significantly increase the specificity of recognition of dying pancreatic acinar cells.
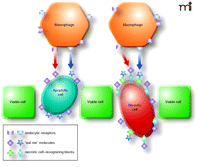
The scheme of clearance of a pancreatic necrotic cell labeled with synthetic “eat-me” marks. Each synthetic molecule consists of “eat-me” fragments (cube and star), such as phospho-L-serine and calreticulin, and the block that recognizes the acinar cells dying by necrosis (cross and diamond). Note that the apoptotic cell externalizes the endogenous engulfment signaling molecules. The receptors for these “eat-me” molecules (callout and chevron) are coded with the same colors as their ligands.
Table 1 (and supplementary data) lists proteins that are annotated in the literature as expressed relatively specifically in the pancreatic acinar cells and which possibly can be used for specific recognition of these cells. Apart from these types of recognizing molecules, the α2,6-sialylated lactosaminic-chain-reactive lectin from Polyporus squamosus mushroom and the agglutinin from Sambicus nigra (elderberry) (SNA), which specifically bind the surface of necrotic cells (32), can serve as nearly ideal recognition blocks, in the context of AP.
Proteins Expressed by Pancreatic Acinar Cells Possibly Useful as Specific Acinar Cell Markersa
Because the engulfment signals can act synergistically (as shown for PS and CRT), the use of several “eat-me” signals linked with antibodies may dramatically improve the cell specificity of the treatment when designed against membrane proteins not fully restricted to pancreatic acinar cells or against secreted pancreatic proteins. In principle, the suggested strategy can also be applied against other acute sterile inflammatory conditions and, possibly, against tumors [for which this idea was originally suggested (49, 50)]. In fact, potentially, the acute inflammatory conditions can be targeted by this approach more directly. In a situation with excessive tissue inflammation, fast apoptotic and necrotic cell removal is a priority. Therefore, as long as “eat-me” signals can be introduced specifically to the dying cells, the anti-inflammatory effect should increase with rising concentration of these molecular signals. The situation may be more complex in cancers (23). Commonly, there is a substantial heterogeneity in the level of expression of different markers within a given population of cancer cells. The head-on approach of labeling tumor cells for quick destruction should, in principle, reduce the tumor mass; however, it is unlikely to eradicate the cancer cell population completely. Therefore, ideally, the designed attack on tumor should leave an immunological memory message. With few exceptions, cell death by apoptosis does not induce an immune response. Finally, insufficient immunogenic stimuli may promote tolerance rather than immunity against cancer cells. Therefore, the choice of “eat-me” signals and their dosage would have to be under much tighter control for cancer treatment than if applied against inflammatory conditions (50, 52).
In summary, the degree of inflammation in the pancreas is determined by the extent of cell necrosis. During AP, the efficiency with which dying cells are cleared by macrophages and the pancreatic stellate cells is likely to be compromised, which may induce further inflammation. The labeling of stressed and, in particular, necrotic pancreatic acinar cells by apoptotic “eat-me” signaling molecules for destruction by phagocytes may be poised to become an effective pharmacological (and essentially anti-inflammatory) approach in the treatment of the acute pancreatitis.
Acknowledgments
This work was supported by the Medical Research Council (MRC) program grant [Grant G0700167].
- Copyright © 2010
References

Alexei V. Tepikin, PhD, is a Professor of Physiology in School of Biomedical Sciences at The University of Liverpool, UK. E-mail A.Tepikin{at}liv.ac.uk; fax +44 (0)151 7945327.

Ole H. Petersen, CBE, FRCP, FMedSci, FRS, is a Medical Research Council Professor and is a Director of The Cardiff School of Biosciences, UK. E-mail PetersenOH{at}cardiff.ac.uk; fax: +44 (0)29 2087 4116.

Michael Chvanov, PhD, is a Research Fellow at The University of Liverpool, UK. E-mail chvanov{at}liv.ac.uk; fax +44 (0)151 7945327.

