Mechanisms of Pathogenesis in Drug Hepatotoxicity Putting the Stress on Mitochondria
- 1 Dept of Medicine, Emory University, 4131 Rollins Research Center, Atlanta, GA 30322
- 2 Dept of Pharmaceutical Sciences, 280 Calhoun St, MUSC, Charleston, SC 29425
- 3 Keck School of Medicine, 2011 Zonal Ave, Los Angeles, CA 90033
- 4 University of Connecticut School of Pharmacy, 69 N Eagleville Rd, Storrs, CT 06269
Abstract
Mitochondria play key roles in aerobic life and in cell death. Thus, interference of normal mitochondrial function impairs cellular energy and lipid metabolism and leads to the unleashing of mediators of cell death. The role of mitochondria in cell death due to drug hepatotoxicity has been receiving renewed attention and it is therefore timely to assess the current status of this area.
Introduction
Mitochondrial impairment is usually a final event common to pathways leading to necrotic and apoptotic cell death. Therefore, it is important to consider whether drug-induced participation of mitochondria in hepatocellular death is a direct result of drugs acting on these organelles (e.g., drug accumulation, inhibition of electron transport and fatty acid oxidation, or depletion of anti-oxidant defense) or an indirect result ensuing from mitochondrial participation in programs of cell death. Whether these programs lead to necrosis or apoptosis, they are mediated through signaling mechanisms arising at the cell membrane (e.g., death receptors) or in subcellular compartments (e.g., the endoplasmic reticulum or cell nucleus). Evidence for a combination of direct and indirect models in drug toxicity have recently emerged, such that a direct effect of drug or drug metabolite promotes mitochondrial production of reactive oxygen species (ROS), whereas an indirect effect occurs upon the evocation of signal transduction programs that culminate in further loss of mitochondrial function.
In this review Dean Jones describes some of the basic principles about mitochondrial function, redox regulation, and ROS production; John Lemasters offers an overview of mitochondrial permeability transition as a target and executioner of cell death; Derick Han assesses the interplay of signal transduction and mitochondria in the acetaminophen model of drug-induced liver injury (DILI); and Urs Boelsterli describes the threshold hypothesis that may explain the long latency often seen in idiosyncratic DILI.
Non-Equilibrium States of Thiol–Disulfide Systems
Oxidative stress is often defined as an imbalance of pro-oxidants and antioxidants; however, the finding that thiols [i.e., glutathione (GSH) and cysteine (Cys)] in plasma are not in redox equilibrium with their disulfide products [i.e., respectively, GSSG and CySS] (1, 2) and that their plasma concentrations are substantially displaced from cellular values (3) has significantly altered concepts of oxidative stress (4, 5). For example, the in vivo “balance” of pro-oxidants and antioxidants cannot be defined by any single entity, such as an equilibrium constant, and our growing knowledge of signaling mechanisms indicates that oxidative stress may be better defined as a disruption of redox signaling, rather than as an imbalance of pro-oxidants and antioxidants (4). The failure of large-scale, double-blind interventional trials with free-radical scavenging antioxidants may likewise reflect an oversimplified therapeutic approach. Thiol–disulfide couples represent key nodes of redox signaling and regulatory mechanisms. Sulfhydryl-containing regulators (e.g., thioredoxins and glutathione) of electron transfer rates can be thought of as biological redox switches and are essential to toxicologic and pathologic mechanisms. The dynamics of non-equilibrium control of redox elements in biologic systems remain a major challenge to our understanding of redox signaling and oxidative stress (6).
An important aspect of redox biology lies in the structures of lipid-containing membranes and multiprotein complexes that function to organize compatible thiol/disulfide couples and orchestrate appropriate redox reactions. The steady-state redox potentials (Eh) of thioredoxin-1, thioredoxin-2, GSH/GSSG, and Cys/CySS couples in mitochondria, nuclei, and secretory complexes, as well as in the extracellular space, are maintained at distinct, disequilibrium values (7). The most highly reducing milieu, promoting the highest rates of electron transfer, is compartmentalized within the mitochondria, which are thus most sensitive to physiologically induced oxidation. In a sense, mitochondria have evolved as compartments that specialize in redox chemistry, and this specialization can make mitochondria selectively vulnerable in pathologic mechanisms.
Mitochondrial Redox Circuits and Thiol-Based Antioxidant Systems
Two types of redox circuits have been distinguished in mitochondria, based upon relative rates of electron transfer (Figure 1) (8). High-flux circuits, in which electron transfer rates are a significant fraction of the rate of cellular O2 consumption, include pathways of intermediary metabolism that maintain energy supply. The high-flux circuits are principally responsible for maintenance of the mitochondrial membrane potential and the production of “high-energy” chemicals (e.g., ATP and succinyl CoA) and effective reductants (e.g., NADPH); high-flux circuits thus subserve biosynthetic and detoxification purposes. In contrast, there appear to be a number of low-flux circuits that subserve a regulatory function and are operationally defined as pathways normally having electron transfer rates less than 1% the rate of cellular O2 consumption. The existence of these low-flux circuits has been largely obscured by the concept that mitochondria generate ROS [e.g., superoxide anion (O2•−) and hydrogen peroxide (H2O2)] as a toxic byproduct of aerobic existence. In fact, O2•− and H2O2 appear to be produced by mitochondria regardless of aerobic conditions and may act as essential signaling molecules to coordinate respiratory functions (8). Superoxide, produced under conditions of ample NADH, inhibits aconitase, a key enzyme for controlling NADH generation, and thereby provides a feedback mechanism to control electron flow. At the same time, superoxide activates uncoupling proteins and thereby prevents excessive membrane potential. Superoxide is also a precursor for H2O2, which appears to be a substrate for several mitochondrial proteins, including GSH peroxidase-(GPX)-1 and GPX4, peroxiredoxin-(Prx)-3, Prx5, thioredoxin reductase-2, and forms of GSH transferase (7, 9, 10); in addition, H2O2 is released by mitochondria and acts as a nuclear signal of NF-κB, Nrf2 and PPAR pathways (10–12). (See Figure 1.)
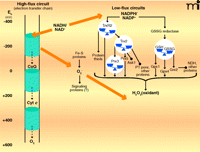
High-flux and low-flux systems of electron transfer pathways. High-flux systems (left), which support ATP production, transfer electrons into the inner membrane electron transfer chain via NADH and electron-carrying proteins such as Coenzyme Q (CoQ) and cytochrome c (Cyt c). The electron transfer to flavin occurs as an electron pair, but the electron transfer chain comprises single-electron transfers until the terminal reduction of molecular O2 to produce two molecules of H2O. At least ninety-nine percent of electron flow in the electron transfer chain results in the reduction of molecular O2. A small fraction of the total flux, probably ≤1% under most physiologic conditions, results in the single-electron reduction of molecular O2 to yield superoxide anion radical (O2•−). The superoxide anion is rapidly dismutated to H2O2 and O2, and the potential role of superoxide as a signaling molecule, rather than merely as unavoidable byproduct of aerobic respiration, remains under investigation. Certain low-flux pathways, which function in signaling and cell regulation, are well established (right), although many more are likely to be elucidated. One of the major NADPH-dependent pathways involves thioredoxin reductase-2 (TrxR2), thioredoxin-2 (Trx2), and the peroxiredoxins-3 and -5. This pathway depends upon H2O2 as an oxidant and regulates apoptosis by means of signal-regulating kinase-1 (Ask1) and the permeability transition (PT) pore. Another major NADPH-dependent pathway involves the mitochondrial isoform of GSSG reductase, GSH, GSH peroxidase-1 and -4, and glutaredoxin-2 (Grx2). Grx2 controls S-glutathionylation of NADH dehydrogenase (NDH) and other proteins. H2O2 and lipid peroxides also function in oxidation of the GSH pathway (not shown). Some iron-sulfur proteins (e.g., aconitase) are also directly oxidized by O2•− The low-flux pathways have been extensively studied as mechanisms to protect against oxidative stress. The fraction of total electron flow diverted to NADPH to support detoxification has not been firmly established. [Approximate scale for steady-state redox potentials (Eh) is indicated at the far left; the thiol/disulfide systems operate between the potential of the NADPH/NADP+ couple (−400 mV) and that of the H2O2/H2O couple (approx +100 mV).]
Mitochondrial GSH and Thioredoxin-2: Parallel, Non-Redundant Signaling Systems
Mitochondria contain two central thiol antioxidant systems, one of which is dependent upon GSH, and the second of which is dependent on thioredoxin-2-(Trx2). Mitochondria cannot synthesize GSH, but rather rely on the dicarboxylate carrier (DIC) and the oxoglutarate carrier (OGC) to obtain GSH from the cytoplasm (in kidney), and at least one other transporter is likely in liver (10). Under many toxicologic conditions, cell death is associated with depletion of mitochondrial GSH and not with loss of cytoplasmic GSH (13). One experimental means for depleting the mitochondrial pool of GSH is to treat cells with the chemical oxidant tert-butylhydroperoxide (tBH), which induces apoptosis and loss of mitochondrial membrane potential. Cells that overexpress the mitochondrion-specific thioredoxin Trx2, however, have been found to be resistant to tBH-induced loss of mitochondrial membrane potential and apoptosis, although mitochondrial GSH undergoes oxidation upon the tBH treatment (14–16). Thus, the antioxidant functions of Trx2 are not redundant, but are rather complementary. Mutations of two essential Cys residues of Trx2 (i.e., C90S and C93S) render the protein ineffective in protecting against tBH-induced cytotoxicity. The C93S mutant form of Trx2 also has a dominant negative character (17). Interestingly, cells overexpressing a Trx2 mutant that lacks an essential Cys manifest enhanced sensitivity to N-ethylmaleimide (NEM), suggesting that certain toxicants function by neutralizing an essential thiol function of Trx2 and essentially converting the protein to a dominant negative form (17).
Transfection studies with the Trx2 C93S mutant show that the protein forms a mixed disulfide with mitochondrial peroxiredoxin-3 (Prx3) but not with peroxiredoxin-5 (Prx5) (18). Expression of the C93S mutant enhances the sensitivity of cells to tBH as well as tumor necrosis factor-α (TNF-α). In cells depleted of GSH by treatment with buthionine sulfoximine (BSO), endogenous Trx2 becomes oxidized and overexpression of Trx2 protects against toxicity, whereas expression of the Trx2 C93S mutant potentiates BSO toxicity. These data again show that Trx2 controls the mitochondrial redox status independent of GSH and is, in cultured neuronal cells, a key component of the defensive mechanism against oxidative stress. The additive protective effects of Trx2 and GSH show that the Trx2 and GSH systems are both functionally important at conditions of low oxidative stress.
In addition to preferential oxidation of mitochondrial Trx2 in response to TNF-α, the magnitude of oxidation of mitochondrial Trx2 (>60 mV) in response to arsenic, cadmium, and mercury, is greater than the magnitude of effect on cytoplasmic Trx1 (20 to 40 mV) (19). This oxidation, along with concomitant activation of apoptosis signal-regulating kinase-1 (ASK-1), occurs with little oxidation of the cellular GSH pool, suggesting that the role of the thioredoxin thiol/disulfide redox couples represents a level of control in apoptotic and toxic signaling pathways that is distinct from mechanisms of control by the GSH/GSSG redox system. This interpretation is also supported by studies showing that Trx2 protects against both the mitochondrial permeability transition (MPT) (20) and the activation of mitochondria-associated ASK-1 (21) (discussed below). Other studies also show that these processes are sensitive to nutrition (22) and that energy and/or substrate supply can contribute to sensitivity of mitochondrial systems to oxidative damage (23).
Extracellular Signals of Oxidative Stress: Mitochondrial Relay in Disease
Mitochondrial Trx2 responds to changes in the extracellular redox potential of Cys/CySS (EhCys/CySS) over a range that is relevant to cardiovascular disease in humans. The availability of Trx2-transgenic mice has been very useful for translation of basic science observations to relevant mouse models (21, 24). Our recent studies with these mice suggest that a signaling pathway from the blood plasma to mitochondria plays a central role in linking oxidative stress and proinflammatory signaling (25). The redox potential of the plasma Cys/CySS couple is oxidized in association with risk factors for cardiovascular disease, including age, smoking, type 2 diabetes, obesity, and alcohol abuse (9, 26). Previous in vitro findings support a cause–effect relationship for plasma CySS in cell signaling pathways associated with cardiovascular disease, including NF-κB-signaling, which controls monocyte adhesion to endothelial cells (27, 28). The overexpression of Trx2 in transgenic mice prevents the increase in mitochondrial ROS that is normally evoked by extracellular plasma CySS as a signal of oxidative stress. Mass spectrometry-based redox proteomics show that several classes of plasma membrane and cytoskeletal proteins involved in inflammation respond to this redox switch, including vascular cell adhesion molecule, integrins, actin, and several Ras family GTPases. Other studies show that proinflammatory cytokines are increased in response to oxidized plasma CySS (12, 29). Together, the data show that the proinflammatory effects of oxidized plasma CySS are due to a pathway that transduces signals from the cell membrane to mitochondria, and that resultant mitochondria-derived oxidative signals activate downstream effector proteins.
Mitochondrial Permeability Transition and Iron in Liver Injury
Chelatable Iron, Oxidative Stress, and Cell Death
Superoxide anion and hydrogen peroxide are among the ROS that promote hepatic injury in response to ischemia-reperfusion, oxidative stress, and many toxic chemicals (Figure 2) (30–33). In the presence of iron, O2•− and H2O2 react to form toxic hydroxyl radicals (OH•) (34). Similarly, iron catalyzes lipid peroxidation chain reactions. Superoxide anion also reacts at diffusion-limited rates with nitric oxide (NO•), a reactive nitrogen species (RNS) produced by nitric oxide synthase (NOS), to form peroxynitrite (OONO−), which in turn decomposes to an OH• -like species. Peroxynitrite also causes nitrosation of tyrosyl residues in proteins (Figure 2A). Both ROS and RNS are important intermediates of toxicity to liver and other tissues (35).
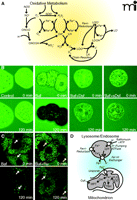
Chelatable iron mobilization and ROS formation. In panel A, oxidative metabolism causes formation of O2•− (i.e., superoxide anion) and H2O2. Superoxide dismutase (SOD) converts some O2•− to H2O2. In the presence of chelatable iron, O2 reduces Fe3+ to Fe2+, and Fe2+ reacts with H2O2 to form hydroxyl radical (OH•). Ferrireductases may also reduce Fe3+ to Fe2+. Hydroxyl radicals react with lipids to form alkyl radicals (L·) to initiate an oxygen-dependent chain reaction generating lipid peroxides (LOOH) and peroxyl radicals (LOO·). Iron also catalyzes a chain reaction generating alkoxyl radicals (LO·) and more LOO·. Nitric oxide synthase (NOS) catalyzes formation of NO•, which reacts with O2•− to form ONOO−. ONOO− decomposes to nitrogen dioxide (NO2·) and OH •. In panel B, the cytosol of mouse hepatocytes was loaded with calcein, and the cells were incubated in culture medium containing calce-in free acid with no further addition (Control), bafilomycin (Baf), bafilomycin plus desferal (Baf+Dsf) or bafilomycin plus starch-desferal (sBaf+Dsf). Note decreased cytosolic calcein fluorescence after bafilomycin, which was blocked by desferal and starch-desferal (see text for details). In panel C, mitochondria and lysosomes of rat hepatocytes were loaded with calcein, and the cells were exposed to bafilomycin (Baf) or bafilomycin in the presence of Ru360 (Baf+Ru), an inhibitor of the mitochondrial calcium uniporter. Note quenching of mitochondrial calcein fluorescence that was suppressed by Ru360. Arrows identify representative lysosomes whose fluorescence increased rather than decreased after bafilomycin (see text for details). In panel D, ferrireductase reduces Fe3+ to Fe2+ in the lysosomal/endosomal compartment. Bafilomycin, as well as ATP deficiency, inhibits the vacuolar proton-pumping ATPase to cause luminal alkalinization and Fe2+ release by apparent Fe2+-H+ exchange. Fe2+ released to the cytosol is taken up by mitochondria via the calcium uniporter as a first hit promoting injury. Mitochondrial oxidative stress by tBH - provides a second hit of O2•− and H2O2 production, and OH• is formed by the Fenton reaction. OH• leads to onset of the CsA-sensitive MPT that causes mitochondrial swelling and uncoupling of oxidative phosphorylation (see text for details).
Iron exists in two pools within cell and tissues. The first of these is “non-chelatable” iron, which resides in ferritin, hemosiderin, and iron-containing prosthetic groups of proteins (e.g., heme and iron-sulfur complexes). Except for enzyme catalysis, this iron is generally unreactive for bulk solution oxidation-reduction chemistry and remains unavailable for chelation by iron chelators like desferal. The second pool comprises free and loosely bound, “chelatable” iron; in fact, Fe2+ and Fe3+ generally associate with polyanionic metabolites like citrate, so that very little chelatable iron is actually free iron. In hepatocytes, chelatable iron concentration is estimated to be about 5 μM (36).
Excessive iron is toxic. An acute overdose causes hepatocellular necrosis, and hereditary hemochromatosis can result in hepatic fibrosis (37, 38). Iron overburden also appears to exacerbate cardiovascular disease, diabetes, cancer, and alcoholic and nonalcoholic steatohepatitis (37, 39–42). In models of oxidative stress, hypoxia/ischemia, and drug-induced liver injury, increased intracellular chelatable iron promotes cell death, whereas iron chelators like desferal are protective (43–50). Such observations imply a role for iron in these injuries, most likely by catalyzing ROS formation. The catalase mimetic TAA-1/Fe blocks iron-dependent cytotoxicity, whereas H2O2 potentiates iron toxicity (51,52), suggesting that toxicity may be mediated by Fe2+-dependent OH• formation from H2O2. In classical iron-catalyzed Fenton chemistry, O2•− regenerates Fe2+ by reducing Fe3+ formed by the reaction with H2O2. However, a permeable superoxide dismutase (SOD) mimetic, MnTBAP, which converts O2•− to O2 and H2O2, fails to protect (53). This finding suggests that a ferrireductase directly reduces Fe3+ to Fe2+, an event that bypasses a requirement for O2•− (Figure 2).
Cellular Uptake of Iron
Transferrin in extracellular fluids delivers Fe3+ to the endosomal/lysosomal compartment by receptor-mediated endocytosis (54). Iron is then released from this compartment for biosynthesis of iron-containing proteins (55). Divalent metal transporter-1 (DMT1) mediates H+/Fe2+ symport in enterocytes and has been proposed to mediate iron release from lysosomes and late endosomes (55, 56). More recent data suggests that type IV mucolipidosis-associated protein (TRPML1) is responsible for release of Fe2+ and other trace metals (e.g., Mn2+ and Zn2+) into the cytosol (57). DMT1 and TRPML1 are specific for Fe2+, and thus endosomal iron release requires intralysosomal reduction of Fe3+ to Fe2+. The endosomal ferrireductase involved is the product of the sixtransmembrane epithelial antigen of the prostate 3 (Steap3) gene (58).
Mitochondrial Permeability Transition
In the mitochondrial permeability transition (MPT), high-conductance permeability transition (PT) pores open that make the mitochondrial inner membrane nonselectively permeable to all solutes of molecular mass up to approximately 1500 Da (59, 60). Calcium ion, oxidative stress, and numerous reactive chemicals induce onset of the MPT, whereas cyclosporin A (CsA) and pH less than 7 inhibit pore opening. After MPT onset, mitochondrial depolarize and undergo large-amplitude swelling driven by colloid osmotic forces, which are the hallmarks of the MPT. Swelling leads to rupture of the mitochondrial outer membrane and release of proapoptotic cytochrome c and other factors from the intermembrane space.
The composition of PT pores is poorly understood and controversial. In one model, PT pores are comprised of the adenine nucleotide transporter (ANT) from the inner membrane, the voltage dependent anion channel (VDAC) from the outer membrane, cyclophilin D (CypD) from the matrix, and possibly other proteins [reviewed in (61, 62)]. However, genetic knockout studies show that the MPT still occurs in mitochondria that are deficient in ANT and VDAC (63–65). Moreover, although CypD underlies PT pore inhibition by cyclosporin A, a cyclosporin A-insensitive MPT still occurs in CypD-deficient mitochondria (66, 67). An alternative model is that PT pores arise from misfolded mitochondrial membrane proteins damaged by oxidative stress and other stresses. Such misfolded proteins aggregate at hydrophilic surfaces thrust into the lipid bilayer to form hydrated aqueous channels. CypD and other molecular chaperones would act to close these channels until opened by specific stimuli, such as high levels of Ca2+ (68). Anion transporters like ANT and the phosphate and aspartate/glutamate carriers seem to be particularly vulnerable to damage and readily misfold to support PT pore formation (69–72).
Release of Chelatable Iron after Lysosomal/Endosomal Compartment Alkalinization
Ferrous (Fe2+) but not ferric (Fe3+) iron quenches the green fluorescence of calcein, which can be used to visualize changes of chelatable Fe2+ (73, 74). In cells that are loaded with calcein and then placed under conditions of oxidative stress, hypoxia, ischemia-reperfusion, or acetaminophen toxicity, a decline in green fluorescence is observed that precedes cell death (75–77). The membrane-permeable iron chelator dipyridyl can restore fluorescence, and desferal (also an iron chelator) protects against cytotoxicity (46, 78–80). Overall, these observations signify that oxidative stress, hypoxia, and acetaminophen intoxication produce an increase of chelatable Fe2+. The source of Fe2+ appears to be the lysosomal/endosomal compartment, as lysosomes are observed to rupture during the course of hypoxia/ischemia, oxidative stress, acetaminophen toxicity and instances of hepatocellular apoptosis (75, 81, 82). The chelatable iron that is released in this way then contributes to iron-dependent ROS formation, MPT onset, and cell death.
Bafilomycin inhibits the vacuolar (endosomal and lysosomal) proton-pumping ATPase, leading to vacuolar alkalinization. In calcein-loaded hepatocytes, bafilomycin treatment raises cytosolic levels of Fe2+ (as indicated by quenching of green fluorescence) over the course of tens of minutes, an effect blocked nearly completely by desferal (Figure 2B) (83). The magnitude of the bafilomycin-mediated increase of Fe2+ is significant (approximating 300 μM if we assume a 1-to-1 stoichiometry of Fe2+ binding to calcein) (Figure 2B). Desferal that is conjugated to starch, and therefore rendered membrane-impermeable, also prevents calcein quenching after the bafilomycin treatment (Figure 2B) (83). Given that starch-desferal is taken up by endocytosis into the lysosomal-endosomal compartment (75), its ability to prevent the bafilomycin-mediated increase in cytosolic Fe2+ implicates lysosomes/endosomes as the likely source of the chelatable Fe2+ that shows up in the cytosol of cells treated with bafilomycin alone (Figure 2B)..
Chelatable Iron and Cytotoxicity after Oxidative Stress and Drug Toxicity
Hepatocytes are generally resistant to oxidative stress induced at low doses of tBH (59, 83). Similarly, bafilomycin alone does not cause cytotoxicity; however, combination treatment with tBH plus bafilomycin results in substantial cell killing, accompanied by elevated ROS production and onset of the MPT. Desferal and starch-desferal prevent this cytotoxicity and block enhanced ROS production and MPT onset (83). Such results illustrate that mobilization of chelatable iron from lysosomes/endosomes potentiates oxidant stress–induced cytotoxicity.
Similarly, mobilization of chelatable iron contributes to acetaminophen hepatotoxicity. In mouse hepatocytes, acetaminophen causes progressive lysosomal rupture and an increase in cytosolic Fe2+ (as indicated by quenching of calcein fluorescence) that is followed by mitochondrial depolarization and onset of the MPT (77). Desferal and starch-desferal prevent the acetaminophen-induced increase in cytosolic Fe2+ and mitochondrial depolarization. These results are consistent with the conclusion that mobilization of Fe2+ from lysosomes/endosomes contributes to acetaminophen cytotoxicity by triggering the MPT.
Iron Uptake by Mitochondria
Calcein can be loaded selectively into mitochondria and lysosomes (76, 84) so that chelatable Fe2+ levels can be monitored according to its respective compartmentalization. Over the course of bafilomycin treatment as discussed above, mitochondrial calcein fluorescence becomes quenched, whereas calcein fluorescence in lysosomes increases (Figure 2C) (83), signifying an increase in mitochondrial Fe2+ and a decrease in lysosomal Fe2+. Both desferal and starch-desferal suppress the bafilomycin-induced rise in mitochondrial Fe2+. The pathway of Fe2+ entry into mitochondria appears to be the calcium uniporter, because Ru360 (85), a highly specific inhibitor of the mitochondrial calcium uniporter, Ru360, also blocks the bafilomycin-induced increase in mitochondrial Fe2+ (Figure 2C), and the uniporter catalyzes uptake of Fe2+ but not Fe3+ in isolated liver mitochondria (86). The molecular identity of the mitochondrial calcium uniporter remains elusive, despite its discovery more than a half century ago. Recently, mitoferrins 1 and 2 were identified as mediating mitochondrial iron uptake (87, 88). It remains to be determined whether mitoferrin 1/2 is, in fact, the mitochondrial calcium uniporter or if the mitoferrins represent an independent higher-affinity iron uptake pathway into mitochondria.
Iron mobilization to Mitochondria as a Second “Hit” in Oxidative Stress and Drug-Induced Hepatotoxicity
Ferrous iron (Fe2+) catalyzes the formation the OH• from H2O2 to promote lipid peroxidation and other deleterious effects (34). After bafilomycin treatment, however, the resulting increase of chelatable Fe2+ is not sufficient to enhance ROS formation, the MPT, and cell death. Similarly, mild oxidative stress is insufficient to produce cytotoxicity. Rather, two “hits” of oxidant stress and increased chelatable Fe2+ are necessary (Figure 2D). The subcellular location of H2O2 and chelatable iron is also important. During oxidative stress and ischemia-reperfusion, hydroperoxide formation occurs primarily within mitochondria (46, 78). Thus, to augment OH• formation, Fe2+ must be transported into mitochondria. In this way, the two required hits—mitochondrial H2O2 generation and uptake of chelatable iron—are provided, promoting lipid peroxidation, the MPT, and other deleterious effects, leading ultimately to cell death (Figure 2D). Overall, the mobilization of chelatable iron from lysosomes to mitochondria is an important factor in hepatocellular injury associated with oxidative stress and drug toxicity.
Interplay of Signal Transduction and Mitochondria in the Acetaminophen model
Acetaminophen is the leading cause of DILI in the US (89). In addition, acetaminophen-induced liver injury in animals provides a valuable model for studying the molecular mechanisms of DILI in humans. The interplay between signal transduction pathways and mitochondria that mediates acetaminophen-induced liver injury can be broken down into discrete steps, including two hits to mitochondria: 1) an upstream hit to mitochondria that initiates mitochondrial injury and increased ROS generation; 2) the activation of signal transduction pathways in the cytoplasm by ROS generated by mitochondria; and 3) a downstream hit to mitochondria caused by JNK and GSK-3β translocation that leads to MPT and hepatocyte death (Figure 3) (90–92).
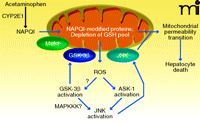
Interplay of signal transduction and mitochondria in acetamin-ophen-induced liver injury. Acetaminophen is metabolized by CYP2E1 to produce the reactive intermediate NAPQI, which modifies mitochondrial proteins and depletes mitochondrial GSH (first hit). The latter leads to increased ROS production, which activates ASK-1, thereby leading to JNK activation. Mitochondrial ROS may also be involved in GSK-3β activation, which in turn helps activate JNK, possibly through MAPKKK. Once activated, both GSK-3β and JNK translocate to mitochondria, thereby providing a sustained second hit. Mcl-1, an important anti-apoptotic protein in mitochondrial membranes, is phosphorylated by GSK-3β, which promotes its degradation. The translocation of GSK-3β and JNK to mitochondria promotes the mitochondrial permeability transition pore opening, full collapse of mitochondrial function, and hepatocyte death. See text for details.
The Upstream Hit to Mitochondria
The first hit to mitochondria in this model is mediated by N-acetyl-p-benzo-quinoneimine (NAPQI), the reactive metabolite of acetaminophen. Acetaminophen is converted by cytochrome P450 (CYP) isoform 2E1 (CYP2E1) to NAPQI, which covalently binds to protein thiols and depletes GSH in the cytoplasm and mitochondria (93). At hepatotoxic doses of acetaminophen (~ 200–300 mg/kg), depletion of GSH and covalent binding by NAPQI cause the partial inhibition of mitochondrial respiration (94, 95) and promote the production of ROS by mitochondria (91). However, this level of mitochondria inhibition is insufficient to induce MPT or cause hepatocyte death. The loss of GSH caused by NAPQI is likely an important factor in causing increased ROS production from mitochondria, because GSH is an essential cofactor for GSH peroxidase, which detoxifies H2O2 in the mitochondrial matrix (96).
Mitochondria in Signal Transduction: JNK and GSK-3β
ROS generated by mitochondria activate signal transduction pathways in the cytoplasm, and the many signaling pathways that are activated in liver following acetaminophen treatment include JNK, glycogen synthase kinase 3β (GSK-3β), and Akt (i.e., protein kinase B) (92). ROS generated from mitochondria are responsible for the activation of JNK in the cytoplasm, and may also be involved in the activation of GSK-3β. The activation of JNK by mitochondrial ROS probably involves ASK-1 (i.e., MAPKKK). ASK-1 associates with reduced thioredoxin, and this association inhibits ASK-1 activity, which provides the basis for ASK-1 activation by ROS (97). Thioredoxin is oxidized by ROS, resulting in its disassociation from ASK-1 and leading to ASK-1 self-activation. Once activated, ASK-1 phosphorylates and activates MKK 4/7, which then phosphorylates and activates JNK. The importance of ASK-1 in acetaminophen hepatotoxicity was supported in recent studies that demonstrated that knocking out ASK-1 blunted JNK activation and prevented acetaminophen-induced liver injury (98). However, ASK-1 is not the only factor involved in JNK activation following acetaminophen treatment, as ASK-1 knockout studies indicate the existence of an ASK-1–independent phase (~1.5 h) of JNK activation in addition to the ASK-1 dependent phase (~3 h). The ASK-1–independent phase of JNK activation following acet-aminophen treatment may be mediated by GSK-3β. Specifically, we have observed that GSK-3β is phosphorylated at an early time point (~1h) following acetaminophen treatment (92). Although phosphorylation of GSK-3β can either reduce or increase its activity (i.e., phosphorylation of either Ser9 or Tyr216, respectively), the observation of an increase in the phosphorylation of glycogen synthase, the downstream target of GSK-3β, suggests an overall increase in GSK-3β activity following acetaminophen treatment. The antisense silencing of GSK-3β delays (~4 h) and blunts acetaminophen-induced JNK activation in liver. GSK-3β modulation of JNK activity may be mediated by mixed lineage kinase (MLK) and mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 1 (MEKK1), both of which have been shown to be regulated by GSK-3β (99, 100).
The Downstream Hit to Mitochondria
Following their activation at hepatotoxic doses of acetaminophen, both JNK and GSK-3β have been shown to translocate to mitochondria and promote hepatocyte death (90, 92). Indeed, the translocation of kinases such as JNK and GSK-3β to mitochondria is a growing theme in many pathologies. In the ischemia-reperfusion heart model, GSK-3β has been shown to translocate to mitochondria and bind to voltage-dependent anion channels (VDAC) and/or myeloid cell leukemia sequence (Mcl-1) to regulate MPT and promote cell death (101–103). JNK translocation to mitochondria promotes release of apoptotic factors, such as cytochrome c, Smac, and AIF, in several cell models following various stresses (104–106). We demonstrated that active purified JNK inhibits respiration in mitochondria isolated from mice after treatment with acetaminophen plus a JNK inhibitor (mitochondria with GSH depleted and covalent binding but no JNK translocation) (91). However, the addition of purified active JNK to mitochondria isolated from control mice had no effect on mitochondrial respiration, suggesting JNK only affects redox-modified mitochondria. The inhibitory effect of active JNK on mitochondrial bioenergetics in redox-modified mitochondria is prevented by co-treatment with cyclosporine A, an inhibitor of MPT. We thus suggest that JNK may inhibit mitochondrial respiration in redox-modified mitochondria by inducing MPT. The mechanism by which JNK modulates redox-modified mitochondria has not been characterized, but bcl-xl and bax are some known downstream targets of JNK in mitochondria (107). The importance of GSK-3β translocation (as opposed to JNK translocation) in mitochondria during acetaminophen–induced liver injury has not been fully determined. GSK-3β translocation promotes degradation of Mcl-1, an important anti-apoptotic protein in mitochondrial membranes (92). The degradation of mcl-1 may alter the balance of pro- and anti-apoptotic proteins in mitochondria in favor of eliciting MPT. Acetaminophen hepatotoxicty is associated with both the loss of antiapoptotic factors (Mcl-1 degradation, bcl-xl and bcl-2 phosphorylation and inactivation) and an increase in proapoptotic proteins in mitochondria (bax translocation) (90, 92, 107). More studies are needed to determine whether GSK-3β translocation to mitochondria is essential in acetaminophen-induced liver injury and whether GSK-3β works with JNK to modulate mitochondria bioenergetics and MPT.
The Shifting View of Drug Hepatotoxicity
The complex interplay between mitochondria and signal trans-duction pathways that involve JNK and GSK-3β is essential in mediating acetaminophen-induced liver injury. Traditionally, acetaminophen-induced liver injury had been viewed as a passive process that ensued after overwhelming injury caused by NAPQI. It is now becoming clear that acetaminophen hepatotoxicity is an active process, depending on specific signaling molecules that interact with mitochondrial functions. The activation of JNK and GSK-3β in inducing hepatocyte death reflects dynamics that extend beyond the direct depletion of GSH as a consequence of acetaminophen metabolism. Because mitochondrial dysfunction and oxidative stress are important components of liver injury caused by many drugs, elucidation of the signal transduction pathways that affect mitochondria will likely be relevant in a diversity of clinical settings.
The Threshold Hypothesis of Mitochondrial Participation in Idiosyncratic DILI
One of the most striking and puzzling clinical hallmarks of idiosyncratic (host-dependent) DILI is the delayed onset of the disease. In fact, the time between initiation of daily drug treatment and the presentation of biochemical markers and clinical symptoms of liver injury can vary from a few weeks to several months, sometimes even exceeding a year (89). The reason for the long lag, often followed by an abrupt progression to DILI, is currently not known. However, it is clear, for the vast majority of drugs, that the delayed time to onset is not related to a gradual accumulation (of drug or drug metabolite) that would eventually lead to critical and toxicologically relevant concentrations in the liver. Instead, the lag time could be explained by an accumulating effect of a drug. This notion, together with experimental findings, is in line with the concept that mitochondria are involved in the etiology of DILI, because damage to mitochondria often reflects successive chemical insults, such that no immediate cause for functional changes or pathological alterations can be established. There is indeed experimental evidence that prolonged injury to mitochondria, such as that which typifies oxidative injury to mitochondrial DNA or to components of the electron transport chain (ETC), has to cross a certain threshold (or a number of thresholds) before cell damage or cell death becomes manifest (Figure 4A).
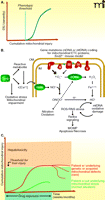
Thresholds for the precipitation of idiosyncratic drug-induced liver injury (DILI) in susceptible patients. The molecular mechanisms that contribute to idiosyncratic DILI can be considered in terms of oxidative injury to mitochondria. A. Hypothetical relationship between cumulative mitochondrial injury and manifestation of liver injury. The dotted line (threshold) represents the “no-apparent-effect level.” (See text for details.) B. Sources and consequences of oxidative/nitrative stress in hepatic mitochondria. Upon deficiencies in components of the electron transport chain (ETC), or by experimentally decreasing the expression of superoxide dismutase-2 (SOD2), the steady-state levels of superoxide anion radical will rise, leading to inactivation of sensitive proteins, such as aconitase or other iron-sulfur cluster-containing proteins (ETC subunits). Alternatively, increased levels of nitric oxide (NO) will readily produce peroxynitrite (ONOO−), which can compromise sensitive proteins including aconitase, complex I, SOD2, and cytochrome c; ONOO− can also produce ultra-reactive hydroxyl radicals. Certain drugs associated with DILI can enhance mitochondrial stress through a number of modes, including an increase in cytosolic (free) Ca2+ levels, which among other actions can activate mitochondrial nitric oxide synthase (NOS). Oxidative/nitrative stress in mitochondria can damage mitochondrial DNA, initiating a catastrophic cycle of dysfunction as abnormal expression of ETC subunits results in further oxidative damage. (MOMP, mitochondrial outer membrane permeabilization; OM, outer membrane; IM, inner membrane.) C. Hypothetical scheme depicting the differential exposure-toxic response relationship against the threshold for inducing overt liver injury. The concept implies that, in normal individuals, drug-mediated injury to mitochondria is mild and does not reach the threshold for liver injury (lower curve; see text for details). In contrast, genetic and environmental factors may impair mitochondrial function, so that the drug-related cumulative mitochondrial injury is more severe, crossing the threshold to overt hepatocellular injury, and DILI may ensue (upper curve). The dotted lines represent possible resolution of the injury after discontinuation of drug exposure.
Threshold Effects in Mitochondrial Toxicity
A number of efficient safety mechanisms are in place within mitochondria to protect cells from high rates of drug-induced mutation, protein oxidation, and the potentially dire consequences of mitochondrial dysfunction per se (108). For example, at the functional level, decreases in expression of certain subunits of ETC complexes do not evoke commensurate decreases in ETC activity and ATP production, unless a certain threshold is reached. Studies with brain mitochondria have revealed that severe impairment in ATP synthesis and oxygen consumption stemming from decreased activity in respiratory complexes I, III, and IV require certain thresholds of inhibition [approximately 25%, 80%, and 70%, inhibition, respectively (109)]. Furthermore, there are also thresholds in place at the genetic level; for example, approximately 60% of mitochondrial DNA must be deleted from the mouse genome before complex IV activity is compromised and serum levels of lactate are elevated (110). This non-linear response can be explained upon consideration that the molecules that subserve mitochondrial function (e.g., mitochondrial DNA, mRNA, and ETC proteins) are present in excess of amounts required for normal cell function. This reserve (or buffering) capacity acts as a protective mechanism; however, at a certain stage of damage, the supply of biomolecules needed to support wild-type mitochondrial function becomes compromised (108). In addition, the hepatocyte harbors thousands of mitochondria that are in a dynamic equilibrium between fission and fusion, which is totally independent of the cell cycle. Oxidatively damaged mitochondria, if not eliminated, can be superseded by proliferation of wild-type mitochondria.
Although mitochondrial threshold effects can plausibly explain tolerance and delayed onset of DILI in susceptible patients, evidence for this mode of action has been rather circumstantial. To provide the proof of concept and investigate molecular mechanisms, novel experimental models are urgently needed for analysis of threshold effects and mitochondrial participation in idiosyncratic DILI. Although the threshold to overt liver injury is not crossed in the vast majority of patients, we have recently hypothesized that underlying mitochondrial abnormalities (genetic or acquired) in certain patients might combine with a superimposed drug stress, leading to overt DILI (111). One approach to model this concept is the utilization of animal models in which mitochondrial function is compromised (112, 113). Ideally, the mitochondrial dysfunction in such animals should be mild and not “clinically” obvious—in fact, a number of human mitochondrial genetic diseases that are clinically discreet are being diagnosed at unexpected rates (114). Furthermore, because the vast majority of human mitochondrial mutations lead to abnormal electron transport and intramitochondrial oxidant stress (115), we sought to model mild mitochondrial oxidant stress. Towards this end, we recently adopted a heterozygous Sod2 knockout mouse and developed it as a potential model to study the mechanisms of mitochondria-mediated liver injury and the involvement of threshold effects. The purpose of using this genetically modified animal model was not to mimic human polymorphisms in Sod2, but rather to provide an in vivo approach to investigating mild mitochondrial oxidant stress, a general consequence of a wide range of genetic or acquired ETC changes.
Drug-Induced Mitochondrial Changes and Liver Injury in the Sod2+/− Mouse Model
Heterozygous knockout of Sod2, a nuclear gene that encodes the mitochondrion-specific Mn-SOD, generates viable mice that do not readily appear to differ from wild-type animals; however, the knockout mice are more prone to mitochondrial oxidant stress, manifesting increased steady-state levels of superoxide, decreased complex I activity, and age-dependent increases in mitochondrial DNA oxidation (116–118). The genetic background of the knockout mice is crucial; for example, knockout mice generated on a C57BL/6J background additionally feature a spontaneous mutation leading to functional deficiency of nicotinamide nucleotide translocase (NNT) (119), which makes these mice even more vulnerable to oxidant stress because their ability to regenerate reduced glutathione in the mitochondrial matrix is compromised. We have recently compared Sod2+/− mice and wild-type controls for their responsiveness to therapeutically relevant doses of a number of DILI-associated drugs (e.g., nimesulide, troglitazone, flutamide, trovafloxacin, tolcapone). Over a 28-day period, we analyzed the time-dependent effects of these drugs on mitochondrial function. We found two major consequences. First, all the investigated drugs invariably decreased the activity of key mitochondrial proteins that are sensitive to oxidant stress (e.g., aconitase-2, complex I) and often decreased the expression of mitochondrial (but not nuclear) genes (120). Second, we found that these markers of mitochondrial injury became apparent only after four weeks, although a number of cytoprotective pathways were activated within two weeks. It thus appears that an initial adaptive response was followed by a toxic response (121), possibly also involving a threshold. Interestingly, these biomarkers were specific for DILI-associated drugs, as a number of bioisosteric analogs that are not associated with liver injury in patients failed to elicit significant mitochondrial oxidative injury.
Mechanistic Studies of Drug-Induced Mitochondrial Injury in the Sod2+/− Model
Increased steady-state levels of mitochondrial superoxide, arising from reduction of Sod2 activity in the Sod+/−mice (i.e., approximately half the wild-type activity), may be exacerbated by drugs that directly target the ETC [e.g., the complex I inhibitors flutamide and troglitazone (122)]. The increased amount of superoxide raises two considerations. First, superoxide that escapes dismutation to hydrogen peroxide cannot cross the inner mitochondrial membrane and can oxidize [Fe-S]-containing enzymes (e.g., aconitase and complex I/III subunits). Alternatively, superoxide can rapidly react with mitochondrial nitric oxide (NO) to form peroxynitrite (ONOO−). For example, the fluoroquinolone antibiotic trovafloxacin (TVX), a typical DILI-associated drug, raises steady-state levels of NO in hepatocellular mitochondria (unpublished data). The mechanisms are not known, but TVX also increases cytosolic (non-ferritin-bound) Ca2+, likely activating the Ca2+-dependent mitochondrial NO synthase (123) to produce ONOO−. Peroxynitrite is dangerous for a number of reasons: i) under acidic conditions, it can be degraded to form the extremely reactive hydroxyl radical; ii) it may directly cause the nitration of aconitase, Sod2, and the [Fe-S]-containing subunits of ETC complexes; and iii) it can induce mitochondrial permeabilization (Figure 4B) (124). This superimposed oxidative/nitrative stress could ultimately push the cell across the threshold to observable injury.
The Practical Application of the Threshold Concept
Increased susceptibility to DILI in murine models is based on genetically determined or acquired underlying defects in mitochondrial function, enhancing mitochondrial oxidative and/or nitrative stress that progressively leads to higher levels of drug-induced mitochondrial injury, eventually crossing the threshold for DILI. If this concept can be confirmed in patients (Figure 4C), drug-induced mitochondrial injury (e.g., compromised aconitase activity, rather than overt liver injury) could be used as a bio-marker to predict the potential hepatic liability of new drugs.
Acknowledgments
DH is supported by a Zumberge award (Office of the Provost, University of Southern California) and the National Institutes of Heath [Grant AA016911]. JLL is supported in part by the National Institutes of Health [Grants DK037034, DK070195, DK070844, DK073336, AA016011]. DPJ is supported by the National Institutes of Health [Grants ES011195, ES009047, ES016731]. UAB is supported by research grants from Pfizer, Inc. and Helsinn Healthcare S.A. NK and DH are supported by the National Institutes of Health [Grants DK067215, DK48522].
- Copyright © 2010
References

Neil Kaplowitz, MD, is Director of the USC NIDDK-sponsored Research Center for Liver Diseases. He holds two endowed chairs, the Brem Professor of Medicine and the Budnick Chair in Liver Diseases, and is Chief of the Division of Gastrointestinal and Liver Diseases. He is also Professor of Physiology & Biophysics and Pharmacology & Pharmaceutical Sciences at the Keck USC School of Medicine and the USC School of Pharmacy. Address correspondence to NK. E-mail kaplowit{at}usc.edu; fax 323-442-3243.

Urs A. Boelsterli, PhD, completed his doctoral studies in biology at the University of Zurich, Switzerland. He is currently Professor of Toxicology at the University of Connecticut, and he holds the Boehringer Ingelheim Endowed Chair in Mechanistic Toxicology. His major research interests focus on molecular mechanisms of drug-induced liver injury.

Derick Han, PhD, is Assistant Professor of Research Medicine at the Keck USC School of Medicine, having also completed his doctoral studies at USC. His research interests focus on oxidative stress, redox chemistry, and signal transduction pathways involved in liver disease.

John J. Lemasters, MD, PhD, is a Professor in the Department of Biochemistry and Molecular Biology and Pharmaceutical Sciences at the Medical University of South Carolina. He is also Director of the Center for Cell Death, Injury and Regeneration at MUSC. His research interests concern the cellular and molecular mechanisms underlying hypoxic and toxic injury to liver and heart cells and organs stored for transplantation surgery.

Dean P. Jones, PhD, is a Professor in the Department of Medicine (Pulmonary Division) at Emory University in Atlanta, GA. He received a PhD in Biochemistry from Oregon Health Sciences University and subsequently carried out research both in the US and abroad. His central research focus is on redox mechanisms of oxidative stress. He currently directs the Emory Clinical Biomarkers Laboratory, which is focused on oxidative stress bio-markers.

