Pharmacology of Store-operated Calcium Channels
Abstract
Store-operated calcium entry is a process by which the depletion of calcium from the endoplasmic reticulum activates calcium influx across the plasma membrane. In the past few years, the major players in this pathway have been identified. STIM1 and STIM2 function as calcium sensors in the endoplasmic reticulum and can interact with and activate plasma membrane channels comprised of Orai1, Orai2, or Orai3 subunits. This review discusses recent advances in our understanding of this widespread signaling mechanism as well as the mechanisms by which a number of interesting pharmacological agents modify it.
Introduction
Calcium signaling is one of the most widespread signaling systems transducing stimuli at the cell surface to modifications of specific cellular functions. The concentration of calcium ion in the resting cytoplasm (i.e., [Ca2+]i) in most mammalian cells is of the order of one or two hundred nanomolar, while extracellular calcium ion concentration is maintained in the low millimolar range. Thus, small perturbations in this large gradient can produce rapid and large increases in intracellular [Ca2+]i. Calcium-responsive proteins, of which calmodulin serves as a relatively ubiquitous example, respond to changes in [Ca2+]i and can evoke a myriad of cellular responses, ranging from the extremely rapid responses of neurosecretory cells and skeletal muscle to the long-term responses involving modulation of cell division and differentiation.
Calcium signals arise from either of two general sources. They result from the release of stored calcium ions within the cytoplasm or from the influx of calcium ions across the plasma membrane, or more typically, from a combination of the two. Pharmacologists have long been interested in calcium signaling because of the link between calcium ions and a number of important clinical targets (e.g., cardiovascular function and inflammation). Calcium ion fluxes across membranes can be triggered either by biophysical processes, such as the gating of voltage-activated calcium channels in the heart, or by ligand-mediated processes, such as the activation of the phospholipase C pathway. In the latter case, signaling is initiated by the action of inositol 1,4,5-trisphosphate (IP3), a second messenger that causes release of calcium ions stored in the endoplasmic or sarcoplasmic reticulum (ER). Activation of phospholipase C also leads to the opening of plasma membrane channels, typically through the capacitative, or store-operated, pathway (1, 2).
In just the past five years, since publication of a review article on this topic (3), we have learned the key molecular components of store-operated channels. This new knowledge has greatly facilitated our understanding of their potential for pharmacological modulation, both by direct channel inhibition and by interfering with their regulation.
Store-operated Calcium Channels
The concept of store-operated, or capacitative, calcium entry into cells simply signifies that changes in the calcium ion concentration in the ER are linked in reciprocal fashion to the rate at which calcium ions cross the plasma membrane and enter the cell (2). The most common physiological mechanism by which calcium ion stores within the cell are released into the cytoplasm is through the action of IP3 downstream of PLC-coupled receptor activation. Experimentally, extensive store depletion can be evoked by reagents such as ionomycin, a calcium ionophore, or thapsigargin, an inhibitor of the ER calcium ion pump (4, 3).
Two major approaches are generally employed for the study of store-operated calcium ion channels, each with particular advantages and disadvantages. Changes in [Ca2+]i can be monitored by use of fluorescent indicators (4). These may be either the BAPTA-type chelating indicators (5) or genetically encoded FRET-based indicators (6); both provide measurement of the concentration of calcium ions in the cytoplasm (or sometimes in intracellular organelles), so that rates of Ca2+ movement across the plasma membrane may be inferred from the change in [Ca2+]i following a step increase in the concentration of external calcium ions (Figure 1A). It is common in such experiments simply to measure the maximum elevation of [Ca2+]i when calcium is (re-)introduced to the extracellular medium, but a more reliable metric may be provided by monitoring the initial rate of the rise of [Ca2+]i.
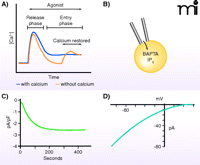
Measuring store-operated calcium entry and Icrac. A) Receptor agonists and Ca2+-depleting agents (e.g., thapsigargin) induce a biphasic increase in cytoplasmic Ca2+, most clearly revealed by protocols involving discharge of Ca2+ stores in the absence of extracellular Ca2+ (dotted line) followed by restoration of extracellular Ca2+. B) The current underlying store-operated Ca2+ entry, Icrac, is generally measured by means of the whole-cell patch-clamp technique, utilizing IP3 and a calcium chelator in the patch pipet solution to deplete Ca2+ stores. C) Depletion of Ca2+ stores while measuring whole-cell currents reveals a relatively slowly developing inward current, Icrac. Depending on the recording configuration and cell types, these currents are typically small, in the range of 0.5 – 3.0 pA/pF. D) Icrac shows strong inward rectification in the current–voltage relationship, as is expected for a Ca2+-selective channel as intracellular Ca2+ concentrations are extremely low.
Fluorescent indicators offer a minimally invasive method of monitoring calcium ion movement across the plasma membrane, but their reliability can be compromised when other factors, such as changes in membrane potential or calcium transporter activity, are at play within the cell. In the face of such interfering activities, the patch-clamp technique, whereby fluxes of calcium ions across the membrane are measured as electrical current, may be preferable. In addition, measurement of calcium ion current permits quantitative analysis of specific biophysical properties, such as selectivity, single channel conductance, and kinetics of activation and inactivation, which collectively can serve as a specific signature for a particular channel type.
The most extensively studied and documented store-operated current is the calcium release–activated-calcium (CRAC) current, or Icrac (7); less extensively investigated currents, with varying (usually lesser) selectivity for calcium have been discussed in recent reviews (1, 8, 9). The single channel conductance of CRAC channels is too low to be measured directly but has been estimated from noise analysis to be of the order of a few fS (10). As a result, Icrac is most commonly studied in a whole-cell configuration to measure total transmembrane current (Figure 1B–D). The invasiveness of this approach poses a disadvantage, as soluble cytoplasmic constituents that may influence channel activity can be lost. In addition, measurement of Icrac is inherently less sensitive than ion movement as detected by fluorescent indicators. Thus, the detection of Icrac, even in cell types with relatively large currents (mast cells, T-lymphocytes), requires great care to reduce calcium feedbacks on channel activity.
The simplest interpretation of the function of store-operated calcium channels is that they assure a relatively constant concentration of calcium in the ER, which is essential for proper protein synthesis and folding and for IP3-induced calcium signaling. However, there is increasing evidence, in at least some instances, that store-operated channels may directly link calcium ion influx to the activation of important downstream response pathways (11–14).
For well over a decade, research into store-operated calcium entry focused on the possible role of the transient receptor potential (TRP) channels, especially the subfamily (i.e., Canonical-type) known as TRPC (15, 16). Despite a large number of research studies, the role of TRPC channels in store-operated entry remains controversial (9, 17). However, in just the past four years or so, RNAi screens have identified two families of small proteins that respectively provide the two primary molecular components of the store-operated calcium entry pathway (8, 18). The first of the two protein families, which functions in sensing calcium ion levels in the ER, comprises the stromal interacting molecules-(STIM)-1 and -2. STIM proteins are single transmembrane proteins that reside in the ER; their N termini are oriented toward the lumen and contain an EF-hand calcium binding motif (19). Depletion of calcium from the ER causes calcium ions to dissociate from STIM, resulting in a conformational change that promotes the self-association and migration of STIM molecules (20) to closely apposed ER–plasma membrane junctions. At these junctions, STIM oligomers interact with proteins that represent the second of the two families (i.e., the Orai family) that have proven essential to store-operated calcium entry. This interaction culminates in the activation of CRAC channels, consisting of pore-forming Orai subunits (Orai1, -2, or -3) (21–27). The interaction of STIM with Orai involves a specific domain in the C-terminal cytoplasmic region of the STIM molecule (28–31) that interacts with specific domains in the N and C termini of Orai (32). The domain structures of STIM1 and Orai1, as well as their modes of interaction in the Ca2+ signaling pathway, are shown in Figure 2. In resting cells, Orai channels are dispersed within the plasma membrane; upon depletion of calcium stores the Orai channels can be seen to aggregate in the vicinity of the sites of STIM aggregation.
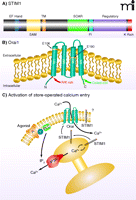
STIM1 and Orai1. A) STIM1 is a single-transmembrane protein localized in the plasma membrane and ER. The N terminus is directed to the lumen of the ER and contains an EF-hand domain that acts as a Ca2+ sensor, followed by a sterile α-motif [(SAM); i.e., a protein interaction domain] and the transmembrane (TM) domain. In the C terminus, within a region of coiled-coil domains, is a STIM–Orai activating region (SOAR), which is involved in activation of Orai channels. Also shown is a region enriched in acidic residues that appears to be involved in fast inactivation (FI) by Ca2+. A regulatory domain contains potential phosphorylation sites that regulate STIM1 function during the cell cycle (61). At the far N terminus is a lysine-rich (K Rich) domain that may contribute to STIM1 localization in near-membrane puncta by interacting with plasma membrane acidic lipids (20). B) Orai channel subunits span the plasma membrane four times, with C and N termini directed to the cytoplasm. Mutations in human Orai1 at positions 106 and 109 alter channel selectivity, providing evidence that Orai proteins are pore-forming units of the CRAC channel. An arginine/lysine rich (R/K Rich) region in the N terminus (99) is involved in coupling Orai to STIM1, as is the coiled-coil C terminus (100). C) Agonism of plasma membrane receptors (R) coupled through a G protein (G) to phospholipase C (PLC) leads to production of IP3, which causes Ca2+ release from the ER through activation of the IP3 receptor (IP3R). The drop in Ca2+ concentration in the ER mobilizes STIM1 to redistribute to near-membrane sites where it activates channels composed of Orai subunits, thereby causing Ca2+ entry. A fraction of cellular STIM1 is also located in the plasma membrane (101) where its function is unclear.
The ectopic expression of STIM1 and Orai1 in transfected cells results in huge Ca2+ signals and huge Icrac-like currents when calcium stores are depleted (22, 23, 27, 33) (Figure 3). This result suggests that STIM1 and Orai1 can fully reconstitute both the signaling and channel functions of store-operated entry. Further, site-directed mutations in acidic residues in transmembrane helix 1-(TM1) or TM3 of Orai1 result in either a complete loss of Icrac or a current with greatly diminished calcium selectivity (34, 35, 26). These results may be considered strong evidence that Orai proteins constitute the pore-forming subunits of the CRAC channel.
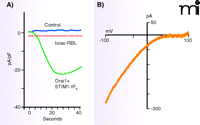
Monster Icrac resulting from overexpressed STIM1 and Orai1. In HEK293 cells overexpressing STIM1 and Orai1, IP3 causes the development of large inward currents with the properties of Icrac. The time course of inward current development is compared to control (A), and the Icrac-like current–voltage relationship is shown (B). Maximum Icrac from the RBL cell line, one of the greatest natural Icrac currents, is shown by the dotted line for comparison. Adapted from (22).
Most, if not all, non-excitable cell types generate calcium ion influx signals upon calcium store depletion; many excitable cell types do so as well. Despite this widespread mechanism of calcium influx, evidence from mouse models as well as humans suggests that this pathway may be amenable to specific targeting through the development of appropriate drugs. Patients that are devoid of either functional Orai1 or STIM1 show severe combined immunodeficiency, primarily associated with a deficit in T cell function (36–39). Generally, these patients die within the first decade of life; however, in a limited number of patients receiving successful bone marrow stem cell transplants, the majority of symptoms disappear (with the notable exception of mildly reduced muscle development).
In Orai1−/− mice, there is diminished innate immunity (due to reduced Icrac in mast cells) (40) as well as impaired musculoskeletal development. There is some disagreement about the involvement of T cells in the mouse models, with one laboratory reporting little or no loss of T cell function (40) and another reporting severe impairment of T cell function (41). The lesser T cell phenotype may have reflected high levels of Orai2 expression in rodents (40). In any event, in both mice and humans, the major phenotype is clearly immunodeficiency. Because these findings result from complete genetic loss of Orai1 store-operated calcium channels or their activator, STIM1, one may speculate that more subtle inhibition might result in highly specific immunosuppression. STIM- and Orai-encoding genes and store-operated entry have been implicated recently in a number of other clinically important situations. For example, STIM1 and Orai have been shown to be important for proliferation of vascular endothelial (42) and smooth muscle (43, 44) cells. A promising link between store-operated entry and breast cancer is also suggested by a recent report showing that knockdown of STIM1 and Orai1 reduces migration and invasiveness of breast cancer cells (45).
Pharmacological Activation of Store-operated Channels
There is as yet only one known small-molecule activator of store-operated channels, namely, 2-aminoethyldiphenyl borinate (2-APB) (46, 47). Although generally described as an inhibitor of store-operated channels (discussed below), the pharmacology of 2-APB is complex. Thus, whereas 2-APB in the 25–100 μM range blocks native store-operated channels, 2-APB in the 1–20 μM range enhances store-operated entry. These responses are reasonably well reproduced in cells overexpressing Orai1 with STIM1 (Figure 4), but different results are seen with Orai2 and Orai3. Orai2 is relatively insensitive to 2-APB (but see below), whereas Orai3 is strongly activated (48–51). Interestingly, when Orai3 is activated by 2-APB, the channel properties are apparently altered, such that the resulting currents show less Ca2+ selectivity. This unique property can serve as a clear pharmacological signature for the expression of Orai3.

Biphasic effect of 2-APB on Icrac.A) During thapsigargin-(TG)-activated store-operated Ca2+ entry in HEK293 cells, addition of 3 or 10 μM 2-APB enhances entry, whereas addition of 30 or 50 μM inhibits. B) Icrac in HEK293 cells overexpressing Orai1 and STIM1 is augmented by 20 μM 2-APB but inhibited by 50 μM. Adapted from (48).
Store-operated channels are of course activated by anything that depletes Ca2+ from the ER, reflecting not a specific pharmacological property of the channels, but having methodological implications for the study of these channels. Experimentally, store-operated currents are most commonly activated following depletion of Ca2+ stores by any of three mechanisms: by activation of the IP3 receptor (as examined by patch-clamp techniques including strong intracellular Ca2+ buffering to reduce Ca2+ feedback); by blockade of SERCA pumps (most often with thapsigargin); or by use of a Ca2+ ionophore (usually ionomycin).
It is not surprising that ionomycin can deplete Ca2+ from the ER and activate store-operated channels; what is a bit surprising, and not well understood, is that within a certain concentration range (<1 μM), ionomycin appears to be specific for intracellular membranes and does not significantly affect permeability of the plasma membrane. General increases in permeability of all cell membranes requires higher (~10 μM) concentrations (52). Activation of Ca2+ entry by the SERCA inhibitor thapsigargin has generally been taken as diagnostic for store-operated Ca2+ entry. However, other non-store-operated channels can sometimes be affected, either positively or negatively, by SERCA inhibitors (17, 53). Likewise, there is evidence that some non-store-operated channels can be activated by IP3 (54). Thus, it is always best to confirm activation of entry by more than one means (e.g., by SERCA inhibition and IP3-mediated signaling).
Two direct activators of store-operated channels have been described. One is a peptide believed to represent the Orai-interacting domain of STIM1 (28–31). Peptides from this region activate store-operated entry, or Icrac, independently of Ca2+ store depletion. A second activator is a mysterious factor called the “calcium influx factor” (CIF) (55). CIF, isolated from store-depleted cells, can activate store-operated Ca2+ entry in the absence of Ca2+ store depletion. It acts independently of STIM1 and is therefore presumed to act downstream of STIM1, implicating the activity of a specific phospholipase A2 (iPLA2) (56). On the other hand, there is credible evidence that STIM1, through its Orai-activating domain, interacts with and gates Orai channels through a direct protein–protein interaction (30). The role of CIF in the store-operated pathway will likely be more critically evaluated once its structure is determined.
Pharmacological Inhibition of Store-operated Channels
With knowledge of the molecular components of the store-operated calcium entry pathway comes the opportunity to develop drugs that either act directly at the pore of the Orai channel (e.g., by blocking the pore) or modulate channel activity (e.g., by targeting STIM1). A number of agents that block store-operated channels, albeit somewhat non-specifically, have been described previously (3, 57, 68). Many of these agents (e.g., inhibitors of cytochrome P450) were suggested to imply something about the mechanism of activation of store-operated channels (3). The discovery of STIM1 has largely superseded many of these theories. Historically, the most widely employed tools for blocking store-operated calcium channels has been the lanthanides Gd3+ and La3+ (3, 4, 59). These trivalent metals completely block Orai channels in the sub-micro-molar range. The only other channels reflecting this degree of sensitivity are the voltage-gated calcium ion channels, which require 1 μM or greater for blockade. The less selective TRPC channels, for example, with which they are sometimes compared, require around 100 μM Gd3+ for complete blockade. The utility of lanthanides is limited in many applications, however, because they must be used in the absence of protein (i.e., serum) and other multivalent anions, the lanthanide salts of which are insoluble.
Within the ER, STIM1 appears to be intimately associated with the microtubule network (60). This association with microtubules appears to facilitate STIM1 function, because disruption of microtubules with drugs such as nocodazole or colchicine partially inhibits STIM1-dependent store-operated entry. The microtubule cytoskeleton is not essential, however, because the inhibition by nocodazole can be fully reversed simply by overexpressing STIM1 (60). During mitosis, when the microtubule cytoskeleton dissociates from the ER to form the mitotic spindle, STIM1 fails to activate store-operated entry. Mitotic suppression of STIM1 function appears to be linked to specific phosphorylation sites in the C terminus (61).
An interesting mechanism-based inhibitor of store-operated Ca2+ entry is the myosin light chain kinase inhibitor ML-9. The ability of this compound to inhibit store-operated entry was originally touted as evidence for a role for myosin light chain kinase in the mechanism of activation of store-operated channels (62, 63). ML-9 blocks the coalescence of STIM1 into puncta and its movement to near plasma membrane sites; ML-9 can even reverse the constitutive puncta produced from EF-hand mutants of STIM1 (64). Surprisingly, the effects of ML-9 do not appear to involve inhibition of myosin light chain kinase (64). The molecular target of ML-9 may be STIM1, but this has not been experimentally demonstrated.
No inhibitor of store operated channels (lanthanides notwithstanding) has received more attention than 2-APB (65). (The ability of 2-APB to transiently activate Orai1 and to produce sustained activation of Orai3 is discussed above.) Because 2-APB had previously been reported to inhibit IP3 receptors (66), its ability to block store-operated entry was interpreted as support for a conformational coupling mechanism involving interaction between CRAC channels and IP3 receptors (67, 68). However, a number of laboratories have demonstrated that neither the activation nor the inhibition of store-operated channels involved IP3 receptors, at least not in a direct way (47, 69–74). In fact, despite its clear ability to inhibit IP3 receptors in broken or permeable cell preparations, 2-APB appears to have little or no effect on IP3 receptors in intact cells (75, 76). In more recent studies, the effects of 2-APB have been studied in the context of the action of STIM1 and specific Orai forms. For example, Orai1-mediated Ca2+ entry is transiently activated, then completely blocked by high concentrations (50 μM) of 2-APB (22, 48, 49). The same appears to be true for Orai2, although activation is limited and the kinetics of block may be slower. Like ML-9, 2-APB inhibits the movement and appearance of STIM1 as puncta at the plasma membrane (48, 77). However, unlike ML-9, this does not likely explain the full inhibitory effect of 2-APB. In cells overexpressing STIM1 and Orai1, 2-APB was considerably less effective in inhibiting STIM1 puncta formation, but was as potent in inhibiting Icrac (48). Thus, from a mechanistic standpoint, the effects of 2-APB on store-operated Ca2+ entry are discouragingly complex.
A series of 2-APB analogs has been synthesized, several of which show substantially increased potency compared to 2-APB (78). Two analogs were investigated in some detail (79). Both DPB162-AE and DPB163-AE inhibited store-operated Ca2+ entry as well as Icrac with a potency almost two orders of magnitude greater than the parent compound. Their potency against the IP3 receptor, however, increased very little, relative to 2-APB, indicating increased specificity. Interestingly, although the two compounds both inhibited Ca2+ entry and Icrac, only one induced the transient activation seen with the parent compound, 2-APB (79). Like 2-APB, both compounds inhibited Icrac through Orai1 channels, partially inhibited Orai2 currents, and induced varying degrees of potentiation of Orai3 currents. However, unlike 2-APB, the selectivity of the Orai3 channels/currents was not diminished. This behavior may reflect the greater size of the analogs, relative to 2-APB, so that they cannot interpolate into the channel pore structure (79).
A small series of pyrazole derivatives has been identified that inhibit NFAT activation and T cell cytokine production (80, 81). The three most interesting were 3,5-bistrifluoromethyl pyrazole derivatives, designated BTP1, BTP2 and BTP3. Of the three, BTP2 was slightly more potent than the other two, blocking IL-2 secretion in the 10–100 nM range (81). The same compound has proven to inhibit store-operated Ca2+ entry in T-cells as well as Icrac (82, 83). Although the concentration dependence of the block of Icrac was similar to that for inhibition of T-cell activation, with an IC50 of about 10 nM, the interpretation of potency is complicated by the fact that the potency of BTP-2 is significantly increased by pre-treatment for periods up to several hours (83). BTP2 may be more specific than other CRAC channel inhibitors because it does not block K+ channels (83) or voltage-gated Ca2+ channels (82); however, BTP2 does block some but not all TRPC cation channels (84).
Although the available evidence suggests that the CRAC channels represent a direct target of BTP2, one report has suggested that this is not the primary underlying mechanism by which it exerts its immunosuppressive effects. Lymphocytes also express a Ca2+-activated non-selective cation channel, TRPM4 (85). TRPM4 functions as a modulator of Ca2+ signaling; Ca2+ signals generated through release of stored Ca2+ or Ca2+ entering cells through CRAC channels activate TRPM4, leading to depolarization and reduced driving force for Ca2+ entry through CRAC channels (86). BTP2 has been reported to activate TRPM4, an effect that would attenuate Ca2+ influx. The potency of BTP2 as a TRPM4 activator was apparently 100 times greater than as an Icrac inhibitor, leading to the conclusion that TRPM4 may be the more relevant target in suppressing T-cell function.
More recently, the actin re-organizing protein drebrin has been identified as a potential site for BTP2 action (87), and knockdown of drebrin by siRNA inhibited sustained Ca2+ signals induced by Ca2+ store depletion. Through an effect on the actin cytoskeleton, the authors of the report speculated, drebrin may play a role in activation of store-operated channels, and BTP2 may act by interfering in this element of the pathway. However, previous studies have generally discounted a major role for the actin cytoskeleton in store-operated Ca2+ entry (88). On the other hand, there is ample evidence for the importance of the actin cytoskeleton in the functioning of TRP channels (53, 89–91), although TRPM4 has not been specifically investigated in this regard.
The availability of recombinant Orai and STIM has resulted in the rapid deployment of high-throughput screens for small-molecule inhibitors of store-operated Ca2+ entry. Because of the particularly strong dependence of lymphocytes on this pathway, the hope is that a relatively specific immunosuppressive drug can be developed. Numerous compounds and series of compounds have appeared in the patent literature, and these are discussed in a recent review by Sweeney and colleagues (92). They are not discussed in detail here because the nature of the screens does not permit much conjecture on the mechanism by which they block store-operated entry, and little information is available as to their specificity. Few have been utilized in studies appearing in peer-reviewed publications. An exception is a Synta compound (Synta 66), used in some recent studies, that is a potent inhibitor of CRAC channels (93, 94); however, its selectivity against a wide panel of ion channels has not been tested. The small molecule inhibitors of store-operated calcium entry discussed in this review are summarized in Table 1.
Small-molecule Inhibitors of Store-operated Calcium Entry
Future Directions
Evidence suggests that STIM1 activates Orai channels by a direct protein–protein interaction across a narrow gap between the ER and plasma membrane (66). Almost simultaneously, four laboratories described sequences from the cytoplasmic C terminus of STIM1 that can directly activate Orai channels independently of Ca2+ store depletion (28–31). A short modulatory domain has also been implicated in fast Ca2+ inactivation of Orai channels (95, 96). This structural information may eventually lead to the rational design of pharmacological agents that can modify STIM/Orai coupling either positively or negatively. Likewise, important pore-lining residues in Orai channels have been identified (26, 34, 35, 97, 98) as well as the region involved in lanthanide blockade (26). Unfortunately, crystallographic structure information concerning Orai channels, STIM1 and STIM2, and their interactions has not been forthcoming. Such information will certainly become available and may reveal additional strategies for pharmacological modification of store-operated Ca2+ entry. Of special interest will be the possibility of inhibitors specific for each of the three Orai isoforms.
Acknowledgments
I thank Stephen Shears, David Armstrong, and Juan Xing for reviewing the manuscript and providing helpful comments. Work from the author’s laboratory described in this review was supported by the Intramural Research Program of the National Institutes of Health .
- Copyright © 2010
References

Jim Putney, PhD, completed his doctoral work in pharmacology at the Medical College of Virginia and then pursued postdoctoral work in the Department of Pharmacology, University of Pennsylvania. He has held faculty positions in the Departments of Pharmacology at Wayne State University and the Medical College of Virginia, Virginia Commonwealth University. Since 1986, he has been Chief of the Calcium Regulation Section, National Institute of Environmental Health Sciences, National Institutes of Health, in Research Triangle Park. He also holds adjunct Professor positions in the Departments of Pharmacology at the University of North Carolina, Chapel Hill, and Virginia Commonwealth University. E-mail putney{at}niehs.nih.gov.

