Molecular Mechanisms of SERT in Platelets: Regulation of Plasma Serotonin Levels
Abstract
The serotonin transporter (SERT) on platelets is a primary mechanism for serotonin (5HT) uptake from the blood plasma. Alteration in plasma 5HT level is associated with a number of cardiovascular diseases and disorders. Therefore, the regulation of the transporter’s activity represents a key mechanism to stabilize the concentration of plasma 5HT. There is a biphasic relationship between plasma 5HT elevation, loss of surface SERT, and depletion of platelet 5HT. Specifically, in platelets, plasma membrane SERT levels and platelet 5HT uptake initially rise as plasma 5HT levels are increased but then fall below normal as the plasma 5HT level continues to rise. Therefore, we propose that elevated plasma 5HT limits its own uptake in platelets by down-regulating SERT as well as modifying the characteristics of SERT partners in the membrane trafficking pathway. This review will summarize current findings regarding the biochemical mechanisms by which elevated 5HT downregulates the expression of SERT on the platelet membrane. Intriguing aspects of this regulation include the intracellular interplay of SERT with the small G protein Rab4 and the concerted 5HT-mediated phosphorylation of vimentin.
Introduction
Serotonin [i.e., 5-hydroxytryptamine (5HT)], an intermediate product of tryptophan metabolism, is primarily located in the enterochromaffin cells of the intestine, the serotoninergic neurons of the brain, and platelets of the blood. 5HT is well-established as a neurotransmitter in the central nervous system (1–4), but it also plays diverse roles in the cardiovascular system, including platelet aggregation and regulation of vascular tone (4, 5). 5HT was discovered by Rapport in 1942 and was isolated from beef serum and named “serotonin” for its vasoconstrictor effect (6). Cardiovascular diseases, including coronary artery disease, atherothrombosis, cerebrovascular ischemia, and myocardial infarction, have been linked to elevated plasma 5HT levels (7–14).
The origins of disease-related elevations in plasma 5HT levels, although controversial, may reflect increased rates of 5HT secretion from enterochromaffin cells of the intestine, which house the majority of the body’s 5HT. But an investigation into the elevation of 5HT levels in the plasma and its relationship to cardiovascular disease—which is our focus here—primarily demands an exploration into platelet biology.
The Serotonin Transporter and Platelet Function
The uptake of 5HT from the plasma and into platelets occurs rapidly, by a saturable mechanism, which makes platelets the fundamental regulators of plasma 5HT concentration. Platelet uptake of 5HT from the plasma is dependent on the serotonin transporter [(SERT); see Figure 1], commonly regarded for its function in neurotransmit-ter reuptake in the central nervous system but also essential to the platelet plasma membrane. After 5HT is transported by SERT across the platelet plasma membrane, it is either sequestered into dense granules by vesicular monoamine transporters (VMAT) or degraded by monoamine oxidase.
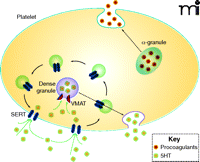
The role of 5HT in platelet function. SERT (dark blue elongated hexagons) within the plasma membrane (light blue line) transports 5HT, which is a signal for platelet activation, from the plasma into the platelet cytoplasm. 5HT is stored within the platelet in dense granules; α-granules store proagulant molecules. Both types of granules can release their contents into the plasma under appropriate platelet stimulation. The internalization and recycling of SERT through platelet trafficking machinery provides a means of regulating 5HT uptake. (See text for details.)
SERT is a member of the Na+/Cl−-dependent solute carrier 6 (SLC6) family, which includes transporters of norepinephrine, dopamine, γ-aminobutyric acid, glycine, proline, creatine, and betaine (15). The detailed mechanism by which SERT activity depends upon transmembrane ion gradients is still not understood; however, the X-ray crystal structure of LeuT, a prokaryotic amino acid transporter and homolog of mammalian neurotransmitter transporters, has significantly elevated our understanding of SERT (16).
A number of groups have attempted to purify SERT to homogeneity, and the protein has been solubilized, through the use of digitonin, in an active form (17–19). Citalopram, a high-affinity ligand for SERT, has been successfully used to create affinity resins and achieve significant purification of the transporter solubilized from platelets and brain tissues (17–19). SERT-encoding cDNA has been isolated (20–22) and sequenced from a number of sources, including human placenta (22), platelets (23), and brain (24); sequences have also been studied from rat (20, 21) and mouse brain (25) and Drosophila (26, 27). The available SERT sequence data show twelve hydrophobic spans connected by hydrophilic loops; both the N and C termini appear to be cytoplasmic (Figure 2). The 630-residue SERT is largely hydrophobic, typifying an integral membrane protein (28). Whereas homo-oligomerization is a common characteristic of transporters for biogenic amine neurotransmitters (29), hetero-oligomeric associations do not appear to be functionally relevant (30).
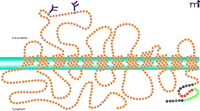
SERT integration into the platelet plasma membrane. Twelve intramembrane domains are predicted for SERT. Sites of glycosylation are indicated at extracellular asparagine residues. The cytoplasmic C-terminal domain contains important regulatory sequences. The SITPET hexapeptide (red residues) is key to SERT trafficking and regulation, putatively providing sites for sequential phosphorylation and interaction with cytoskeletal elements (e.g., vimentin). Residues indicated in green have been implicated in the binding of Rab 4. (See text for details.)
The functional roles of the sialylated N-glycans moieties of SERT have been investigated through mutational analysis. Results of these studies indicate that sialylated glycans modifications are required for homomeric SERT–SERT interactions and are also involved in interactions between SERT and myosin IIa (31). This latter interaction is relevant in the phosphorylation of SERT; SERT is phosphorylated by a cyclic guanosine monophosphate-(cGMP)-dependent protein kinase (PKG) that is anchored at the plasma membrane by myosin. The oligosaccharide chains are also essential to this interaction as well as to the cGMP-dependent stimulation of SERT function. Because the N-glycosylation and myosin-association domains are located at opposite sides of the plasma membrane, we have hypothesized that the glycosyl modification of the extracellular domain imparts favorable conformational effects to the integral protein and allows SERT to participate in functional protein–protein interactions within the cytoplasm (31).
Biphasic Relationship Between 5HT Concentration and Sert Activity
It comes as no surprise that the capacity of platelets and cells to take up 5HT is generally proportional to the number of SERT molecules located in the plasma membrane. But intriguingly, the surface expression of SERT molecules on neurons and glial cells is regulated by the concentration of extracellular 5HT (32, 33). For these cells, the elevation of extracellular 5HT levels functions to decrease the surface expression of transporters, thereby limiting their synaptic availability; this change in surface expression is mediated by changes in the trafficking dynamics of SERT (33–36).
The impact of plasma 5HT concentration on the surface expression of SERT in the platelet plasma membrane has been recognized; however, the nature of this regulation, which is both subtle and complex, is only beginning to come to light in the literature. It is clear that the treatment of platelets with 5HT affects the density of SERT molecules in the plasma membrane (36). Recently, by analyzing the impact of 5HT, at various concentrations (0–2.5 nM), on platelet SERT, we have found that the relationship between the surface expression of SERT in platelets and concentration of 5HT exposure is biphasic (37–39). Specifically, plasma membrane SERT levels and 5HT uptake initially rise as platelets are exposed to increasing 5HT levels, but this initial response is followed by a second phase, whereby higher concentrations of 5HT cause SERT levels to fall below baseline. In vivo and in vitro, these studies have confirmed the interrelationships among extracellular 5HT concentration, surface expression SERT, and depletion of platelet 5HT to be strikingly dynamic. Rats (40) and mice (41) deficient in SERT have also helped to elucidate the relationships among platelet SERT expression, circulating 5HT levels in plasma, and the contribution of these influences to platelet physiology. For example, platelets from SERT knockout rodents are almost completely devoid of 5HT. Nevertheless, knockout models do not provide an opportunity to investigate the effect of altered serotonin levels in the plasma upon the surface expression of SERT in platelets or concomitant physiological responses mediated by SERT.
Platelet SERT Dynamics: Implications for Regulation of Blood Pressure
Several lines of evidence demonstrate that plasma 5HT is directly related to systemic hypertension (37–39, 42–48); its potent vaso-constrictor activity is synergistic with that of epinephrine (49), and certain 5HT antagonists may lower blood pressure (50). Furthermore, hypertension is a salient manifestation of 5HT excess, as reflected in carcinoid syndrome (51) and serotonin syndrome (52). In patients with uncontrolled hypertension, the uptake of 5HT by platelets was significantly impaired as compared to platelets examined after hypertension was controlled (53). Further studies have demonstrated that transcriptional regulation of SERT expression as well as SERT inhibitors (54–58) alter the plasma level of 5HT and induce the development of hypertension. The involvement of SERT in the development of hypertension is additionally of great medical interest, because SERT represents the target of many clinically important drugs such as cocaine, amphetamine, and antidepressants. Regulation of the transporter’s activity could constitute an important mechanism for the control of neurotransmitter action during hypertension.
Blood plasma and platelets isolated from hypertensive individuals are thus of interest in studying the impact of high plasma 5HT concentration on platelet SERT. For example, we have collected blood samples from adult men presenting for emergency care with high blood pressure (trauma- or stress-associated hypertension), and we have analyzed platelet SERT from these patients during and subsequent to symptom presentation (37). We found the plasma concentration of 5HT to be as high as 2 nM from admitting patients and approximately 0.7–1.0 nM after hypertension subsided. Moreover, the 5HT uptake rates that typified hypertensive platelets were low relative to platelets collected under normotensive conditions, and this lower rate of uptake reflected a decrease in Vmax, without any significant effect upon the Km for 5HT. The effect on Vmax reflected a decrease in the density of SERT on the platelet membrane, with no change in whole cell expression. Additionally, the concentration of 5HT within platelets collected under hypertensive conditions was 33% lower, relative to normotensive platelets, whereas the hypertensive plasma concentration of 5HT was increased by 33% (37). Pretreatment of the platelets isolated from normotensive blood samples with 5HT, however, results in the lower level of SERT molecules that typified platelets isolated under hypertensive conditions.
The 5HT uptake rate of 5HT-pretreated platelets is intriguing. 5HT uptake initially rises as plasma 5HT concentration increases but then falls below normal at higher 5HT concentrations (37, 44, 45). Thus, the surface expression of SERT on platelets may be uniquely altered in response to plasma 5HT levels, which in turn changes platelet 5HT content. The decreased surface expression of platelet SERT in the presence of high 5HT concentrations in plasma may function under hypertensive conditions to delimit 5HT uptake, thereby providing a feedback effect. In addition, we found that platelets aggregate more readily following pretreatment with the higher concentration (2 nM) of 5HT.
These observations may seem counterintuitive. When extra-cellular 5HT concentration is increased, the level of SERT on the plasma membrane might well be expected to increase, in order to promote 5HT uptake from plasma. However, the impact of a substrate on the surface expression of the family of Na/Cl-dependent monoamine transporters may resemble the effect of a ligand on the activity of G protein–coupled receptors, whereby ligand binding can initiate not only signal transduction but also endocytosis. Internalization, recycling, and trafficking of receptor tyrosine kinases within the endosome compartment are each regulated to control the overall process of downregulation. For example, the action of brain-derived neurotrophic factor through its receptor has been reported to play critical roles in survival, differentiation, and synaptic activity of neurons (58); Rap2, a member of the Ras GTPase family, modulates the trafficking of receptors for Activin/ Nodal and thereby regulates signaling activity. In the absence of ligand, Rap2 directs internalized Activin/Nodal receptors into a recycling pathway, thereby preventing receptor degradation. However, after ligand activation, receptor recycling by Rap2 is terminated (59). The targeting of SERT by therapeutic agents that enhance serotoninergic signaling (e.g., serotonin-selective reuptake inhibitors) and by drugs of abuse [e.g., cocaine and MDMA (i.e., ectasy)] (60, 61) is known to include effects on recycling and internalization of SERT (33). Substrate-mediated modulation of transporter trafficking and surface functionality is also exemplified by the increased surface expression of the excitatory amino acid transporter (EAAT1) in response to glutamate, as well as the GABA-mediated increase in glutamine transporter 1 (GAT1) activity (62–64). Similarly, the surface expression and activity of the dopamine transporter (DAT) is enhanced by dopamine, amphetamine, and cocaine (65).
As is the case for many membrane proteins, SERT trafficking is mediated by vesicular packing and interactions with specialized proteins. Depending on many factors, such as substrate availability, inhibitors, and interacting proteins, SERT-carrying small vesicles will either reside in the platelet cytoplasm or translocate to the plasma membrane. Upon clearance of 5HT from the extracellular matrix into the cytoplasm of platelets, SERT moves from the plasma membrane and becomes sequestered in small vesicles that may be routed elsewhere. In platelets, the biosynthesis as well as the posttranslational modifications of proteins is minimal. These features make platelets a unique system for evaluating factors that play roles in membrane trafficking, crucial to our attempts to understand the plasma level 5HT–dependence of SERT recycling.
The N- and C-terminal sequences of monoamine transporters have been studied extensively within the context of transport function and localization. The C-terminal region of DAT and NET is very important for transporter function, expression, and localization (66–69). Syntaxin 1A (62, 70), secretory carrier membrane protein 2 (63, 71), Hic-5 (36), and α-synuclein (72), are among the factors that interact with SERT and regulate the translocation of transporter among the cytoplasmic compartments. Furthermore, several proteins have been identified in association with the C terminus of SERT: PICK1 (73–75), the actin cytoskeleton (76), neuronal nitric oxide synthase, Sec23A, Sec24C (73), and fibrinogen, an activator of integrin αIIbβ3 (77).
SERT activity, trafficking, and phosphorylation can also be modulated through C-terminal interaction with a member of the MARCKS family of protein kinase C substrates (78). PKC-mediated phosphorylation of SERT has been correlated with extracellular 5HT levels (36, 39, 79), and the twenty-residue C terminal peptide sequence of SERT is critical for the functional expression of the transporter (39, 80). Importantly, Whitworth and colleagues demonstrated that 5HT-mediated signals have a role in regulating the number of transporters at or near the synapse by changing the subcellular redistribution of SERT in neurons and glia (81). These and other studies confirm the regulatory and functional significance of transporter trafficking, internalization, and recycling (22, 37–39, 71, 82). It should be noted that the posttranslational modification of SERT (e.g., glycosylation) also regulates transporter function (81), but given that glycosylation occurs in megakaryocytes (i.e., the progenitors of platelets), this aspect of SERT regulation may not be altered in platelets.
5HT-Mediated RAB4–SERT Interaction
At high cytoplasmic concentration (12 μM), 5HT accelerates the exocytosis of α-granules by causing the serotonylation and concomitant constitutive activation of small GTPases, such as Rho and Rab4 (83–88). There are more than sixty members in the mammalian Rab protein family, and individual Rab proteins are localized to the cytoplasmic leaflet of distinct compartments in both the endocytotic and exocytotic pathways (89). These proteins are similar in mass and sequence to the yeast YPT1 and SEC4 proteins, which have important functions in endocytosis and exocytosis. Rab4 is associated with early endosomes and regulates membrane recycling (89–91). In adipocytes, Rab4 controls recycling of the insulin-regulated glucose transporter GLUT4 (92), and recombinant Rab4 stimulates secretion of α-granules in platelets (83, 84).
Platelets contain high levels of a Ca2+-dependent transglutaminase that catalyzes a transamidation reaction between 5HT and small GTPases, including Rab4 (i.e., resulting in the serotonylation of Rab4) (83, 84). In response to elevations in plasma 5HT, the platelet phosphatidylinositol pathway is activated, which increases intracellular Ca2+ levels and thereby activates the transglutaminase. In this way, elevations of 5HT concentration in the plasma results in the serotonylation of Rab4 within its phosphate binding domain (83, 84). Interestingly, the analogous serotonylation of Rho results in constitutive activation (93).
We have examined the association of Rab4 and SERT in a heterologous expression system and platelets. In both systems, Rab4 and SERT can be seen (i.e., by coimmunoprecipitation and immunofluorescence) to associate only in the presence of high plasma 5HT (38). More specifically, we found that a C-terminal sequence of SERT, between T616 and D624, was necessary for the interaction with Rab4. Using variants of Rab4 that were either constitutively active or unable to bind nucleotides, we furthermore found that SERT can only associate with the active form of Rab4 (Rab4·GTP), which occurs after the serotonylation of Rab4 (83, 84). However, a constitutively active form of Rab4 was able to bind SERT in the absence of 5HT. Therefore, these findings may indicate the importance of activation of Rab4 independent of 5HT level, as elevated 5HT is but one of the many other factors that can activate Rab proteins. In light of these data, we hypothesize that at high concentrations of 5HT in the blood plasma, 5HT is taken up by platelets at rates that saturate the VMAT capacity of dense granules; the saturation of VMAT results in its inactivation through a G protein–dependent mechanism (94–96). At the same time, the concomitant high level of cytoplasmic 5HT would result in the serotonylation and activation of Rab4, thereby promoting the association between cytoplasmic SERT and Rab4·GTP. In this way, the trafficking of SERT to the plasma membrane would be impeded, and the concomitant reduction in surface expression of platelet SERT would reduce the uptake of 5HT from the plasma (Figure 3).
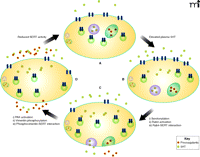
SERT-mediated 5HT uptake: reciprocal regulation of plasma 5HT levels and SERT activity in the platelet plasma membrane. Upon elevation of 5HT concentration (yellow squares) in the plasma (upper right arrow), the uptake rate of 5HT increases, as trafficking vesicles (green circles; see [A] and [B]) deliver SERT (blue geometries) to the platelet surface. The rise in cytoplasmic 5HT [B] promotes the transamination of Rab4 (not schematized), thereby activating Rab4 and promoting its association with SERT; the Rab4–SERT interaction prevents the trafficking of SERT to the plasma membrane [C]. The rise in cytoplasmic 5HT also promotes exocytosis of dense granules (purple circles) and α-granules (green ovals), the latter of which contain procoagulants (hexagons). The internalization of SERT is also promoted as 5HT activates PAK, which phosphorylates vimentin (see [C] and [D]), causing its association with SERT. The indicated roles of Rab4 and vimentin culminate in the reduced surface expression of SERT, which delimits further intake of 5HT, restoring cytoplasmic levels to their initial values (see [D] and [A]). (See text for details.)
Plasma 5HT and Vimentin
Vimentin is a type III intermediate filament involved in cytoplasmic trafficking in cells of mesenchymal (e.g., endothelium, fibroblasts, megakaryocytes) and myogenic origin (97). It is a minor component of the platelet cytoskeleton (98) and resolves to the Triton X-100 insoluble fraction of human platelets (99, 100). Vimentin participates in a network of intermediate filaments that extends beneath the cell membrane and regulates the activity of the p21-activating kinase (PAK), which functions in the transduction of diverse extracelleular signals to alter intracellular pathways (101). Significantly, vimentin is a substrate of PAK, and PAK becomes activated in cells that are exposed to 5HT (101). Following phosphorylation at Ser56, the curved filamentous structure of vimentin undergoes reorganization and straightens (100).
Given its role in the trafficking of proteins, we became interested in vimentin as a possible mediator in the effect of 5HT concentration upon surface expression of SERT. At physiological plasma 5HT levels, the vimentin–SERT association was found at intracellular locations as well as the plasma membrane (45). However, when plasma 5HT concentration was higher than physiological levels, the association between SERT and vimentin was enhanced, corresponding to an altered vimentin network. To investigate whether this association is related with the phosphorylation and the reorganization of vimentin filaments, immunofluorescence of cells expressing SERT was performed before and after 5HT pretreatment (45). This approach clearly illustrated that the filamentous structure of vimentin was straightened, following 5HT pretreatment, in concert with the enhancement of phosphovimentin-SERT association. Moreover, SERT tagged by extracellular biotin and isolated from the platelet plasma membrane is associated with vimentin, suggesting that SERT bridges vimentin to the plasma membrane (45). When the extracellular 5HT level is elevated, PAK is activated and phosphorylates vimentin, which enhances the association between phosphovimentin and the cytoplasmic domain of SERT at the plasma membrane, which leads to reduced density of SERT molecules on the plasma membrane and thereby reduces 5HT uptake (45). Accordingly, in cells overexpressing the S56A mutant form of vimentin (lacking the phosphorylation site), 5HT uptake rates of cells are normalized even in the presence of elevated plasma 5HT levels (45). Thus, under conditions where the blood plasma 5HT level is high, as it is in hypertension, reduced surface expression of platelet SERT [i.e., the basis for the reduction in the Vmax for 5HT uptake (37)] can be linked to PAK signaling and phosphorylation of the vimentin framework.
Indeed, we have determined that the C terminus of SERT is required for binding to vimentin (45). Progressive truncation of the SERT C terminus maps an essential domain for the binding of vimentin to between residues 611 and 616 (i.e., a SITPET sequence; see Figure 2). If this sequence is removed (i.e., a deletion of the twenty residues at the SERT C terminus), the protein is not delivered to the plasma membrane and remains localized in the cytoplasm; if the hexapeptide (i.e., SITPET) is retained as the C terminus (i.e., deletion of fourteen residues relative to the wild-type protein), the resulting construct does not alter 5HT uptake rates, but its association with vimentin is precluded.
A series of site-directed mutations has provided intriguing glimpses into possible basis for the SERT–vimentin interaction and the 5HT-mediated regulation of SERT expression at the platelet surface. The serine and two threonine residues in the essential (vimentin-binding) C-terminal SITPET sequence (see Figure 2), given the regulation of SERT by phosphorylation, have been substituted by alanine (thereby removing potential sites of phosphorylation) and aspartate (thereby providing an irreversible negative charge that could possibly mimic the effect of a phosphate group). Replacement of the three residues with alanine, either simultaneously (i.e., the triple mutation) or one at a time, has no significant effect on 5HT transport activity, vimentin binding, or cellular trafficking of SERT. In contrast, a triple mutation that substitutes aspartate residues for the serine and threonine residues within the SITPET sequence prevents the trafficking of the transporter to the plasma membrane and causes SERT to accumulate at intracellular locations. The accumulation of negative charge at the C terminus of SERT, which would by supposition occur upon phosphorylation of three residues within the SITPET sequence, can be mechanistically related to the reduced surface expression and transport activity of SERT in response to high concentration of 5HT in the plasma.
Additionally, the stepwise mutation of each of the serine and threonine residues within the SITPET sequence proves to be relevant to the finer aspects of SERT regulation. Specifically, the phosphorylation of SERT has been suggested to occur in a two-step process (102), according to which the serine residue is first phosphorylated, thereby inactivating 5HT transport. In the second step, phosphorylation of the threonine residues has been suggested to result in the internalization of SERT. In regard to this two-step hypothesis of phosphorylation functionality, we have found that single mutation of the SITPET serine residue to aspartate reduces transport capacity and SERT surface density by ~39%, with concomitant accumulation of SERT in the cytoplasm (45). SERT binding to vimentin is also particularly sensitive to this particular mutation in SERT.
The double mutation that substitutes aspartic acid in place of both threonine residues within the SITPET sequence, on the other hand, reduces uptake capacity to ~16% of the wild-type rate (45). The mutational data thus indicate additive effects of placing negative charges within the SITPET sequence (to mimic phosphorylation), and we might hypothesize that step-wise phosphorylation sequentially exposes the vimentin-binding domain (i.e., the SITPET sequence) on the C terminus of SERT (45). Our mutational analysis, however, indicating the localization of the Ser611D mutant protein in the cytoplasm, is at odds with the original two-step model.
Conclusion
SERT has a rich history in pharmacology and has been translated into a widely exploited therapeutic target. New insights into the regulation of SERT function and surface expression in platelets may open new dimensions in the targeting of SERT. We have focused here on the regulation of platelet SERT by extracellular 5HT and the relevance of this regulation in hypertension.
Transamination of Rab4 with 5HT places this small GTPase into a constitutively active form that accelerates the exocytosis of α-granules to secrete procoagulants into the plasma (83, 84), binds SERT to block its translocation to the plasma membrane (44), and thereby limits 5HT uptake by platelets. This process may thus contribute to elevated plasma concentrations of 5HT and appears to activate PAK-dependent pathways. Among these pathways, and one that is experimentally implicated in regulation of SERT by plasma 5HT, is the phosphorylation of vimentin, a modification that uncurls the filamentous structure of vimentin, enhances vimentin–SERT association, and accelerates the internalization of SERT from the plasma membrane (45).
Finally, excess 5HT in the plasma may enhance platelet aggregation induced by other endogenous substances; however, even at the highest levels of plasma 5HT, there are always a number of SERT molecules on the platelet membrane that still continue to clear plasma 5HT, albeit at a reduced rate that is presumably maintained as long as the plasma 5HT concentration remains high (37). Thus, a highly dynamic relationship appears to exist between plasma 5HT concentration and SERT trafficking that may be capable of influencing platelet function.
Platelets are derived from the cytoplasm of megakaryocytes and enter the circulatory system in an inactive form. An initial activation of platelets stabilizes them in hemostasis. Further activation and aggregation at fibrin-stabilized hemostatic areas can elaborate into thrombosis, which is a leading cause of death for patients with hypertension and cardiovascular disease. Although 5HT as a therapeutic target is widely recognized in the context of the central nervous system, it is also well established in amplifying platelet activation; once cytoplasmic calcium levels are elevated, the exocytosis of dense granules releases 5HT into the plasma (83, 84). Indeed, in the absence of peripheral 5HT, platelets exhibit a blunted secretion of α-granules and a reduced risk of thrombosis. Conversely, the exocytosis of α-granules is accelerated in the presence of elevated plasma 5HT levels (83, 84) and by concurrent activation of platelets (4, 48, 103–104). However, neither the precise role of 5HT as a signaling molecule in the activation of platelets nor the mechanisms by which 5HT mediates aggregation is yet known. Furthermore, the involvement of SERT in the activation of platelets has not been sufficiently studied in vivo.
Considering the role of 5HT in platelet aggregation, a loss of platelet SERT coupled with elevated plasma 5HT may play a significant role in the cluster of cardiovascular diseases, including diabetes, metabolic syndrome, atherosclerosis, and peripheral arterial disease, which are thought to reflect a prothrombotic state. Numerous factors have been identified that confer susceptibility to thrombosis, including a loss of endothelial-derived nitric oxide, vascular smooth muscle cell hypertrophy, hyperinsulinemia and other metabolic abnormalities, obesity, and inflammation (1, 4, 84, 48, 103–104). The development of possible antithrombotic therapies for patients with cardiovascular disease has focused on reducing these risk factors rather than on promoting endogenous mechanisms of anti-thrombosis. In this regard, therapies designed to promote the expression of SERT on the platelet surface and thereby reduce plasma levels of 5HT may represent a novel approach to alleviating thrombotic events.
Acknowledgments
We thank Samuel Freyaldenhoven for critical review of the manuscript and his participation in helpful discussions related to this work. This work was supported by the American Heart Association [Grant 0660032Z] and by the National Institutes of Health National Heart Lung and Blood Institute [Grants R01HL091196 and R01HL091196-01A2W1] to FK.
- Copyright © 2010
References

Fusun Kilic, PhD, is an Associate Professor and Charles P. Mercado, MD, is a Postdoctoral Fellow in the Department of Biochemistry and Molecular Biology at UAMS, College of Medicine, in Little Rock. Their research focuses on the impact of elevated plasma serotonin on the serotonin transporter. Send correspondence to FK. E-mail kilicfusun{at}uams.edu; fax 501-686-8169.

