Age-related Macular Degeneration: Genetic and Environmental Factors of Disease
- 1 Department of Ophthalmology and Vision Science, Eye and ENT Hospital, Fudan University, Shanghai, 200031, China
- 2 Institute for Genomic Medicine and Shiley Eye Center, University of California at San Diego, La Jolla, CA, 92093-0838
- 3 Department of Ophthalmology, West China Hospital, Sichuan University, 610041, China
Abstract
Age-related macular degeneration (AMD) is the most common cause of visual impairment among the elderly in developed countries, and its prevalence is thus increasing as the population ages; however, treatment options remain limited because the etiology and pathogenesis of AMD are incompletely defined. Recently, much progress has been made in gene discovery and mechanistic studies, which clearly indicate that AMD involves the interaction of multiple genetic and environmental factors. The identification of genes that have a substantial impact on the risk for AMD is not only facilitating the diagnosis and screening of populations at risk but is also elucidating key molecular pathways of pathogenesis. Pharmacogenetic studies of treatment responsiveness among patients with the “wet” form of AMD are increasingly proving to be clinically relevant; pharmacogenetic approaches hold great promise for both identifying patients with the best chance for vision recovery as well as tailoring individualized therapies.
Introduction
Age-related macular degeneration (AMD) is the most common cause of visual impairment among elderly individuals in the developed world, occurring primarily in persons over the age of 50 (1–7). It is estimated that 1.75 million people in the United States suffer from advanced AMD, and 7.3 million people are affected with intermediate AMD, which represents increased risk for development of advanced disease (8). AMD thus poses a major public health problem with significant economic and social impact.
AMD is an abnormality of the retinal pigment epithelium (RPE) that leads to photoreceptor degeneration of the overlying central retina, or macula, and loss of central vision (9–10) (Figure 1). The macula (Figure 2) is 5–6 mm in diameter, and at its center is the fovea, responsible for greatest visual acuity. Early AMD is characterized by subretinal deposits, known as drusen, that measure greater than 60 μm and hyper- or hypo-pigmentation of the RPE. Intermediate AMD is characterized by the accumulation of focal or diffuse drusen measuring greater than 125 μm and hyper- or hypo-pigmentation of the RPE (Figure 2). Advanced AMD can be classified into either of two categories. The first of these comprises geographic atrophy (GA; i.e., dry, or non-exudative, AMD), which is characterized by a sharply delineated area of REP atrophy measuring at least 175 μm along one dimension and including visible choroidal vessels (Figure 2C). The alternative form of the advanced disease is choroidal neovascularization (CNV; i.e., wet, or exudative, AMD), which may involve some or all of the following: subretinal neovascular membranes; subretinal fluid, exudates, and hemorrhages (Figure 2D); pigment epithelial detachment (PED; see Figure 1B); and subretinal/intraretinal scarring. Advanced AMD can result in loss of central visual acuity and lead to severe and permanent visual impairment and blindness. Whereas dry AMD accounts for 80–90% of all cases of advanced disease, more than 90% of AMD patients with severe loss of central vision manifest CNV (11).
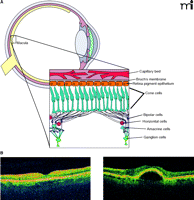
The retina revealed. A. Diagrammatic representation of the retina. Inset shows the several types of neuronal cells within the macula, the retinal pigment epithelium (RPE), and choroid. B. Two optical coherence tomography images of a normal (left) and AMD retina with pigmented epithelium detachment (PED) (right).
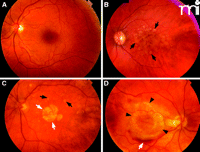
Fundus images from patients representing several AMD subtypes. A. Normal macula. B. Macula with confluent soft drusen (black arrows), a hallmark of early AMD. C. Macula of dry AMD with soft drusen (black arrows) and geographic atrophy GA (white arrows). D. Macula of choroidal neovascularization (CNV) or wet AMD with subretinal hemorrhage (black arrows).
The etiology and pathophysiology of AMD are poorly understood. Drusen, which are the histological markers of AMD, are yellowish extracellular deposits of lipid, protein, lipoprotein, and cellular debris that accumulate between the RPE and Bruch’s membrane or within Bruch’s membrane (12–15). Drusen can include complement components and modulators (4, 16–21) and the serine protease HTRA1 (22). The cause of drusen may be linked to one or more of the following key processes that are implicated in AMD: 1) increased outer segment turnover; 2) impaired activity/function of the RPE; 3) free radical/oxidative damage; 4) aging and degeneration of elements of Bruch’s membrane (e.g., collagen and elastin); 5) reduced clearance of material from Bruch’s membrane into choriocapillaries; and 6) deleterious immune system activation (23). Specific alterations in Bruch’s membrane composition that have been suggested to cause drusen include excessive lipid deposition and protein cross linking as well as impaired permeability to nutrients.
Genetic and Environmental Risk Factors
Although advancing age is the greatest risk factor associated with the development of AMD, environmental and lifestyle factors may significantly affect individual risk. Smoking is an important, modifiable factor that has been consistently associated with a twofold increased risk for developing AMD (odds ratios ranging from 1.8 to 3) (24). Oxidative stress and antioxidant depletion have been implicated in retinal damage from smoking, although the precise mechanism in AMD remains unclear (25–27). Other factors that have been reported to influence risk for AMD include sunlight exposure, alcohol consumption, increased plasma fibrinogen levels, diet, hypertension, body mass index (BMI), and iris color (24, 27–31). Regarding diet, antioxidants such as carotenoids, zinc, and vitamins A and E may provide a protective benefit against AMD.
Epidemiological studies have demonstrated differences in the prevalence of AMD based on ethnicity, with prevalence among Caucasians being greater than that among non-white groups (32–33). Such ethnic differences may reflect genetic as well as environmental risk factors. AMD has a significant genetic component (34–37), as several twin studies have shown significantly higher concordance rates between monozygotic twins as compared to concordance between dizygotic twins (38–42). Familial aggregation studies from general populations (43–44) and tertiary eye care centers (45–46) reveal that family members of individuals with AMD are at increased risk (2.4- to 19.8-fold) for developing the disease relative to individuals with no family history.
Genetic Studies of AMD
Gene association studies reveal that multiple genes may be associated with AMD (Table 1). An association between AMD and the gene that encodes apolipoprotein E (APOE) has been reported, through multiple studies (47–51), such that the ɛ2 allele increases risk, whereas the ɛ4 allele has a protective effect. In a screen for sequence variation among the gene family that encodes the fibulin (FBLN) glycoproteins, Stone and colleagues detected missense mutations in 1.7% of 402 AMD patients (52). Seven different missense mutations of the FBLN5 gene have been discovered in individuals examined from eight families affected by AMD with a distinctive mutation corresponding to each of the affected individuals (53). Evidence from genetic association studies in conjunction with the demonstration of complement deposition in the retina and choroid has implicated toll-like receptor-(TLR)-3 and -4 in the development of certain cases of AMD (53, 54). Most recently, two large genome-wide association studies have established another two susceptibility loci. A functional promoter variant of the hepatic lipase–encoding gene (LIPC) was found to be strongly associated with advanced AMD, corroborating the involvement of lipid pathways in AMD development (55). Additionally, a locus near the gene for metallopeptidase inhibitor 3 (TIMP3), which is involved in degradation of the extracellular matrix, was found to be associated with AMD (56). Other candidate genes include C3, C2/CFB, VEGFA, ABCA4, ERCC6, and CX3CR1 (Table 1).
Genetic Loci Associated with AMD
The most convincing evidence for the genetic contribution to AMD is the identification of major disease susceptibility alleles on chromosomes 1q32 (which includes the gene that encodes complement factor H [CFH]) and chromosome 10q26 (which includes PLEKHA1, hypothetical gene LOC387715, and HTRA1). Specifically, risk of developing AMD is associated with an allele of CFH in which a histidine residue is encoded in place of a tyrosine residue at amino acid position 402 (16, 57, 58). The increased risk ranges between 2- to 4-fold for heterozygote carriers and 3- to 7-fold for homozygotes. In addition, multiple other polymorphisms, many of which are in non-coding regions of CFH or in nearby genes encoding other complement factors, demonstrate equal or stronger association with disease susceptibility than does the CFH Y402H variant (16, 59). Importantly, no single polymorphism could account for the entire contribution of CFH to disease susceptibility. Rather, multiple polymorphisms defined a set of four common haplotypes (two of which promoted disease susceptibility and two of which were seemingly protective) and several comparatively rare haplotypes (all of which were associated with increased disease susceptibility) (60, 61). At 10q26, containing genes PLEKHA1, LOC387715, and HTRA1 (62–65), a single nucleotide polymorphism (SNP) in the promoter of the HTRA1 gene was associated with a population attributable risk of 49.3% and a 10-times greater risk of developing CNV (22, 66).
The preeminence of the HTRA1 SNP in accounting for the association of 10q26 to disease was subsequently established also in a Utah population; however, LOC387715 manifested very high association as well, suggesting that the AMD risk with regard to 10q26 may, similar to the scenario in the CFH region, reflect linkage as a function of alternative haplotypes. When disease-associated alleles exist within both the HTRA1 and CFH loci, the population attributable risk for AMD is estimated to be 71.4% (22).
Pathogenic Mechanisms in AMD
The complement activating genes CFH, CFB, and C2, as well as the serine protease family gene HTRA1, have the strongest associations with AMD and offer a glimpse into possible pathogenic mechanisms. LOC387715 is also highly associated. But because it has no known protein function, its role in pathogenesis cannot be adequately hypothesized. CFH binds and inactivates C3b, an important complement protein, and this process contributes to the selectivity of innate immune responses (67). Domain 7 in CFH has been shown to contain the site of binding specificity that allows CFH to recognize heparin or sialic acid on host cells (68).
The AMD-associated Y402H polymorphism discussed earlier happens to occur within domain 7 of CFH (16), possibly reflecting that the binding specificity of CFH is altered in such a way as to preclude recognition of host cell surface molecules and the corresponding inactivation of bound C3b. Accordingly, the Y402H substitution would leave host cells, particularly RPE and choroid-associated cells, vulnerable to complement-mediated degradation, which is consistent with the observation that CFH is expressed in drusen (16). Whether or not CFH variants result in drusen formation or merely accumulate within drusen is not known; however, with CFH expressed in drusen, its accumulation results in increased risk of RPE and choroidal cell degradation, threatening the overlaying retina. The end result would be photoreceptor degeneration and, ultimately, AMD. Other CFH, CFB, and C2 variants could act through similar pathways.
The AMD-associated promoter polymorphism observed in HTRA1 presumably alters a binding element (recognized by transcription factors AP2 and SRF) and thereby potentiates HTRA1 expression (22, 62). HTRA1 is a serine protease and a key modulator of proteoglycans degradation in the extracellular matrix (69). HTRA1 proteolytic activity permits other degradative enzymes, such as collagenases and matrix metalloproteinases, to access their respective substrates (70). Similar to complement factors, HTRA1 is expressed in drusen of AMD patients (22). Excessive accumulation of HTRA1 in drusen could compromise Bruch’s membrane integrity, thereby allowing expansion of choroidal capillaries and resulting in neovascular AMD. HTRA1 is also a potent inhibitor of transforming growth factor-β, which is involved in extracellular matrix formation and angiogenesis (71) and thus represents another potential pathway to AMD. Alternatively, HTRA1-mediated destabilization of Bruch’s membrane could contribute to RPE atrophy and GA.
Risk Prediction Models
The key to establishing the mechanisms of pathogenesis in AMD will depend on an understanding of the interplay between the genetic and environmental factors that have been implicated in the disease. The development of risk models that consider this interplay facilitates the identification of patients at greatest risk for developing AMD or progressing through the various stages of the disease. Risk models have been developed to predict the prevalence and incidence of AMD by duly considering multiple gene variations [i.e., in CFH (57, 72, 73), HTRA1/LOC387715 (22, 66), C2 (59), CFB (59), and C3 (74, 75)] in concert with environmental, ocular, demographic, behavioral, and treatment factors (76).
Multiplicative models demonstrate that each genetic variant noted above is independently associated with AMD, and when patients carry high-risk variants on both of the major susceptibility genes, the risk of developing AMD increases tremendously (Figure 3). The discrimination accuracy of predictive models is limited to known factors, accounting for only about 40–60% (27, 65, 77–79) of the genetic risk for developing AMD in Caucasians (after adjustment for other genetic and environmental factors). Recently, sensitivity has been improved to 88% in an AMD predictive model through the inclusion of biomarkers of complement components and activation fragments (i.e., Ba, C3a, C3d, C5a, and factor D) (80, 81); however, specificity still remains around 73%. (Here, “sensitivity” is the probability that the model correctly identifies AMD-affected individuals, whereas “specificity” is the probability that the model correctly identifies healthy individuals.) In order to predict the occurrence and progression of AMD precisely enough to be used clinically for diagnosis and prognosis, it is crucial that we identify additional genetic or environmental contributors for incorporation into predictive models.
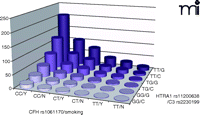
AMD risk profile as a function of disease-associated genes and smoking status. Joint effect of specific alleles of CFH (rs1061170, Y402H), HTRA1 (rs11200638), C3 (rs2230199) and smoking status were shown by odds ratios calculated by a logistic regression model, adjusted for age, gender, and body mass index (BMI).
Early identification of high-risk individuals could allow clinicians to better preserve patient vision through targeted surveillance and therapeutic intervention. High-risk individuals would also benefit from targeted education regarding healthy lifestyles. Smoking is the most important and obvious environmental factor that patients could eliminate to decrease their risk for AMD. Although the combined risk for AMD increases with age, regardless of genetic background, smoking increases the probability of developing AMD by 10–15%. Furthermore, carrying even one risk allele of CFH advances the potential age of disease onset by 10 years, regardless of smoking status (Figure 4).
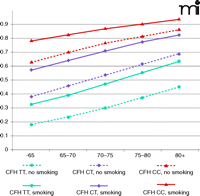
Lifelong risk for developing AMD. Probabilities of developing advanced AMD were estimated by evaluating genotypes of CFH (rs2274170) and smoking status. Low risk, median risk, and high risk associated with CFH genotypes were coded as blue, purple, and red, respectively. Solid lines represent smoking group and dashed lines represent nonsmoking group.
Implications of Genotype in the Treatment of AMD
The etiology and pathophysiology of AMD are becoming increasingly better characterized as the number of genetic factors implicated in the pathogenesis of AMD increases. Understanding the role of genetics in AMD allows both direct and indirect tailoring of therapies to target specific molecules and pathways. However, studies must first prove that therapeutic differences correlate with genotypes before clinical practice recommendations can be changed. Currently, there is a small but growing body of evidence showing that an individual’s genetic profile may influence existing therapies for AMD.
Genotype and Response to Photodynamic Therapy
Photodynamic therapy (PDT) is a treatment for AMD-related CNV. Intravenous administration of the photosensitizer verteporfin, followed by activation with a non-thermal laser, initiates photo-chemical reactions, involving singlet oxygen and reactive oxygen intermediates, that damage endothelial cells lining the CNV; the therapeutic rationale is thus to prevent thrombosis and selective occlusion within CNV vessels. However, the efficacy of PDT treatment is limited. Although several ocular predictive factors, such as baseline visual acuity and CNV size at presentation, have been considered to explain the individually variable efficacy of PDT, they have only weakly contributed to effective optimization of PDT. Evaluations of the potential role of common AMD-associated gene variants in determining CNV responsiveness to PDT have been inconsistent. Brantley and colleagues found that visual acuity in PDT-treated individuals with the CFH TT genotype was significantly worse than that of PDT-treated TC and CC genotypes (82). Goverdhan and colleagues found the degree of visual loss following PDT to be significantly higher in the CFH CC genotype (83). Two other studies (84–85) reported no significant association between CFH genotype and responsiveness to PDT. No effect upon PDT responsiveness has been associated with particular LOC387715 or HTRA1 variants (86). Instead, two SNPs affecting the CRP gene and two SNPs in the VEGF gene (see below) demonstrated significant association with response to PDT (85, 87). Moreover, peculiar polymorphisms in genes that encode coagulation balance factors (e.g., factor V, prothrombin, and factor XIII-A) as well as in the MTHFR gene have been correlated to the degree of post-PDT benefit in Caucasian patients with neovascular AMD (88–90). Although patient cohort size was limited in these retrospective studies, the correlations identify an opportunity to optimize patient eligibility criteria for PDT (Table 2).
Pharmacogenetic Studies of Treatment Outcome in AMD
Genotype and Responsiveness to Anti-VEGF Treatment
Vascular endothelial growth factor (VEGF) is a major molecular mediator of neovascularization and is present in CNV membranes in wet AMD. VEGF inhibitors have been used to successfully treat exudative AMD and have become clinical standards. Ranibizumab is a humanized antibody fragment that targets VEGF-A and all of its biologically active degradation products. Phase 3 clinical trials showed that ranibizumab can stabilize vision in greater than 90% of patients and significantly improve vision over a two-year period in one-quarter to one-third of patients with neovascular AMD; the drug was approved by the FDA for treatment of neovascular AMD in 2006 (91–92). Bevacizumab is an FDA-approved anti–VEGF-A antibody for treatment of metastatic colorectal and breast cancer. Retrospective case series have demonstrated efficacy and safety of off-label bevacizumab for treating neovascular AMD (93). The ongoing National Eye Institute-(NEI)-sponsored comparison of AMD treatment trials will compare ranibizumab with bevacizumab, administered monthly, for the treatment of neovascular AMD in 1200 patients over a two-year period.
An investigation of the effects of CFH and HTRA1/LOC387715 genotypes on the responsiveness of neovascular AMD to intravitreal bevacizumab revealed that visual acuity improved in only 10.5% of treated patients who were homozygous for the CFH Y402H (CC) genotype, whereas visual acuity improved in 53.7% of treated patients who possessed CFH TC and TT genotypes (P = 0.004). In fact, mean visual acuity worsened, from 20/206 to 20/341, among the patient population who possessed the CC genotype (P = 0.016). On the other hand, the LOC387715 genotype did not appear, after adjusting for age, pretreatment visual acuity, and lesion size, to affect patient responsiveness to bevacizumab (P=0.18) (94). Because both bevacizumab and ranibizumab target the same VEGF-A molecules, it is expected that the same pharmacogenetic relationship should exist for ranibizumab. Indeed, a retrospective analysis of 156 patients with exudative AMD who had been treated with intravitreal ranibizumab monotherapy and followed for nine months (95), indicates that patients with the CFH (CC) genotype tended to require more injections of drug than did other patients, which is indicative of a potential pharmacogenetic relationship between CFH genotype and ranibizumab treatment outcome (Table 2).
Genotype and Nutritional Supplementation
In 2001, the Age-Related Eye Disease Study (AREDS), a large, multicenter, double-masked, placebo-controlled clinical trial, established that a combination of zinc and antioxidants (β-carotene, vitamin C, and vitamin E) produced a 25% reduction in development of advanced AMD (over a five-year period) and a 19% reduction in severe vision loss in individuals determined to be at high risk of developing the advanced forms of the disease. Use of these oral supplements has become standard practice in the US and remains the only therapy for intermediate and advanced dry AMD (96–97). The Study also revealed that CFH Y402H genotype could influence the effect of zinc in the supplementation regime. Specifically, the greatest benefit was seen in those individuals with the low-risk CFH genotype, among whom the rate of AMD progression was lowered by approximately two-thirds. In contrast, AREDS-type supplements seem to have less impact on those with the high-risk CFH genotype (98). Moreover, an interaction (P= 0.004) was observed in the AREDS treatment groups taking zinc when compared with the groups taking no zinc, but not in groups taking antioxidants compared with those taking no antioxidants (P = 0.59). These findings suggest that the genotype-treatment interaction in participants with CFH TT genotype may be related primarily to the zinc component of the supplements. Genetic screening could thus help to identify those individuals at high risk for developing advanced AMD and could benefit patients who are the most likely to respond to supplements (Table 2).
Conclusions
Great strides have been made in the past five years in identifying genes associated with AMD, although much work is yet needed to understand the complex molecular genetics of this disease. Future endeavors, building on current scientific successes, should include: 1) detailed phenotyping to more accurately describe populations; 2) large-scale population studies allowing for better interpretation of mixed findings; and 3) functional studies of AMD-associated genes in both in vitro and in vivo systems.
Future studies need to put more emphasis on the harmonization of descriptions (e.g., primary features, age of onset, environmental factors) of AMD cases. Large datasets can then be generated to allow stratification of general phenotypes for genome-wide linkage analysis and association studies into specific phenotypes, such as GA and wet AMD, or primary features, such as soft drusen. There may, for example, be genes that confer risk for GA, or for exudative AMD, or for both together. Certain genes may help determine the subset of early AMD that progresses to advanced AMD. Analysis of quantitative trait loci, which assumes that the genes that confer risk for the late form are the same genes as for the early form, may lose power if such genes are distinct. Further defining subphenotypes with respect to environmental factors could reveal gene-environment interactions that would otherwise go undetected.
Accurate phenotype descriptions, standardized across multiple populations, may lead to high-density marker sets and supply result consistency and statistical power in analyzing further association and linkage studies. Many of the genes in Table 1 have been strongly associated in one or more population studies but not in other populations. High-density genotyping arrays will enhance consistency by providing richer genetic descriptions of populations studied.
Finally, functional studies are necessary to demonstrate that AMD-associated genetic variants can indeed lead to biological changes. Techniques from cell transfection to genetically engineered animals will allow researchers to examine the functional roles of multiple alleles in AMD onset and progression. Genetically engineered animals are ideal for not only understanding the pathophysiology of AMD, but also for discovering possible therapeutic interventions. The ability to reproduce genetic findings in cell and animal studies is a necessary step in attributing true disease association to specific genetic variants.
In this way, the burgeoning field of pharmacogenetics will enable clinicians to tailor pharmacotherapy to a patient’s specific genetic variations. Identification of genetic risk variants will be useful in identifying high-risk populations for early treatment. Examples of diseases for which a genetic profile has already assisted in treatment include statin treatment in cardiovascular disease (99), imatinib treatment for chronic myeloid leukemia (100), and warfarin dosing with respect to cytochrome P450 CYP2C9 genotypes (101). As one of the most well-characterized, late-onset, complex diseases in terms of clinical features and underlying genetic and environmental influences, AMD is an excellent disease to which the principles of personalized medicine can be applied. Early detection of patients with high-risk genotypes and implementation of smoking cessation should significantly reduce the age of onset and severity of AMD. Furthermore, genotype-based customizing of treatment of neovascular AMD by anti-VEGF therapy will allow clinicians to identify optimal responders to maximize treatment benefit and reduce costs and side effects.
Acknowledgments
This work was supported by the National Institutes of Health [Grants R01EY14428, RO1EY18660] . We further wish to acknowledge support from Research to Prevent Blindness, the Ruth and Milton Steinbach Fund, Ronald McDonald House Charities, the Macular Vision Research Foundation, Burroughs Wellcome Fund Clinical Scientist Award in Translational Research, and West China Hospital of Sichuan University.
- Copyright © 2010
References

Yuhong Chen, MD, PhD, is an ophthalmologist at the Eye and ENT Hospital of Fudan University in China. She has been working as a postdoctoral fellow in ocular genetics at UC San Diego since 2008.

Matthew Bedell, MD, graduated this year from the UCSD School of Medicine. He is working as a postdoctoral scholar in ocular genetics and planning on a career in clinical and research ophthalmology.
Kang Zhang, MD, PhD, is a faculty member at Shiley Eye Center and director of the Institute for Genomic Medicine at UCSD. His research mainly focuses on novel disease gene targets and therapies for macular degeneration, glaucoma, diabetic retinopathy, and inherited retinal degenerations. Address correspondence to KZ. E-mail kang.zhang{at}gmail.com; fax 858-246-0873.

