The Promise of Epigenetics in Personalized Medicine
Abstract
Numerous preclinical and clinical trials, with older as well as some newer drugs, have demonstrated the targeting of aberrant epigenetic marks to be a viable means of preventing and treating certain human disorders, including myelodysplastic and leukemic syndromes and various hemoglobinopathies. These findings are encouraging, and although the risks associated with such therapy are largely unknown, precise maps of epigenetic marks are becoming increasingly available through advancements in sequencing protocols that combine chromatin immunoprecipitation and gene expression analyses. Indeed, progress in understanding gene regulation at promoter regions and chromatin organization in health and disease has been substantial. New insights into the proteins that are targeted by therapeutic agents that alter epigenetic programs may provide important inroads into personalized medicine.
Introduction
Human genomic DNA measures approximately 1.7 m in length when fully extended but is folded into a five-μm-wide nucleus in cells in a manner that permits it to be replicated and transcribed (1, 2). Robert Kornberg and his colleagues and other investigators defined the X-ray structure of the basic building block of chromatin, the nucleosome, showing that it consisted of approximately 146–base pairs of DNA wrapped around a histone octamer core particle containing two each of histone H2A, H2B, H3, and H4 (1, 3), or alternatively, histone variants. Each of these DNA-histone cores is linked together by histone H1, resulting in a greatly condensed form that is sufficiently small to fit in the cell nucleus (2, 4). The nucleosome is a dynamic participant in the regulation of virtually all chromosomal processes, including transcription, replication, and DNA repair. Methyl groups serve as epigenetic marks of DNA whereas other moieties (acetyl, phosphoryl, ubiquinylation, etc.) serve as epigenetic marks of core histones of chromatin. Combinations of existing epigenetic marks and functions of specific enzymes and nonhistone coadaptor molecules act to modify and remodel chromatin during gene expression. It is the addition and removal of these marks and remodeling of the nucleosome that give chromatin its dynamic character (5).
The older epigenetic literature focuses mainly on developmental biology (6), but as researchers’ interests broadened and deepened, epigenetics came to encompass both chromosomal and molecular biology (7). Today, epigenetics is frequently described as engaging all heritable changes in gene expression in the absence of changes in the DNA sequence, and currently, it comprises one of the most rapidly expanding fields of biology. Although the study of human disease has focused mainly on genetic mechanisms, currently many human disorders are thought to be driven by combinations of genetic and epigenetic abnormalities. Indeed, hardly a day goes by without the appearance of epigenetic aberrations touching a new or another aspect of human disease (8–10). One would also expect that heritable drug responsiveness would be influenced by epigenetic variation, but so far there have been few reports for this possibility (11–13).
Perhaps the main function of the epigenome—with its capacity to respond with reversible modifications—is to serve as a functional intermediary between the heritable genome (genomic DNA) and the stresses imposed by the environment. The success of the personalized epigenetic approach to therapeutic and preventive measures of human disease depends on a familiarity with the epigenetic modifications that determine the choice of targeted therapies and the capacity of molecular diagnostic tests that enable doctors to predict accurately those patients who can benefit from such therapies.
This review begins by delineating epigenetic maneuvers and mechanisms that accompany DNA methylation and histone modifications found in normal cells, and follows this with a brief summary of how alteration or disruption of epigenetic modifications and patterns cause human cancer. Subsequently, the article inquires as to what extent epigenetically focused tools and concepts are relevant to personalized medicine.
Epigenetic Patterns in Normal Cells
More than 200 types of specialized cells comprise the tissues and organs of the human body. The genetic sequence for every cell type in the body is the same; however, each different cell type has its own characteristic set of marks that define its epigenome. These marks change with developmental stage (14, 15), gender (16), age (16, 17), and in response to environmental chemicals (18–22), including nutrients (23), and stress (24). In normal cells, the patterns of epigenetic marks are “remembered” by the cell so that daughter cells develop and mature according to a program that is similar to that of their parent cells. Because the epigenome amounts to the sum of all cellular epigenotypes, screening for epigenetic marks and abnormal epigenetic regulation is much more difficult than genetic screening.
Regulatory epigenetic mechanisms in normal cells include the effects of DNA methylation at the carbon-5 position of cytosine (Figure 1) on gene expression and chromatin packaging and remodeling through post-translational modifications of core histones, and of regulatory microRNAs on gene expression and epigenotypes. Of these, DNA methylation and histone modifications, particularly those located in promoter regions of genes, are of primary interest to our discussion because these processes have been demonstrated to be potentially therapeutic in reversing or preventing the disease phenotype.
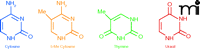
The chemical structures of cytosine, 5-methylcytosine, thymine, and uracil.
DNA Methylation
The discovery of DNA methylation occurred several years before that of post-translational histone modifications. Perhaps not surprisingly, much more has been learned of DNA methylation than of histone modifications.
Methylation of cytosine is a modification of eukaryotic DNA in which a methyl group is enzymatically transferred from S-adenosylmethionine to the 5-position of cytosine. The methyl group is situated in the major groove of the DNA and inhibits transcription by interfering with transcription factor binding proteins but does not interfere with DNA replication or repair. The pattern of genomic methylation is explained by two distinct processes—de novo methylation (rendered by Dnmt3a and Dnmt3b) and maintenance methylation (rendered by Dnmt1) (Figure 2). De novo methylation occurs in the early embryo, and is the enzymatic transfer of the methyl group to unmethylated cytosines of CpG nucleotides, whereas maintenance methylation rapidly converts a “hemimethylated CpG” (a CpG in which only one strand of DNA is methylated) into a symmetrically methylated form. Dnmt3L, another member of the Dnmt3 family, is unusual because it lacks key methyltransferase motifs; however, it retains the capacity to repress transcription.

De novo and maintenance DNA methylation in eukaryotes.
Defining the significance of DNA methylation at CpG residues picked up momentum in the 1980s. Gardiner-Garden and Frommer (25) recognized the importance of regions of high-density methylation in DNA of vertebrates and proposed they be defined as “CpG islands,” while others reported (26) that 35% of mutations causing human disease were located within CpG islands and that these mutations occurred at a frequency 42-fold higher than that predicted from random mutation. Additionally, DNA methylation was shown to prevent damage from transposed elements such as infectious proviruses by suppressing the capacity of such “selfish” foreign elements to disrupt gene structure and function. This DNA damage–preventing action was thought to be the ancestral function of methylation in invertebrates and vertebrates were thought to have retained this function but additionally had adapted DNA methylation at CpG residues as suppressors of endogenous promoters (27–29). Other investigators affirmed these ideas later for large, repetitive genomic regions, including ribosomal DNA, satellite sequences, centromeric repeats, parasitic elements, and endogenous retroviruses (30, 31).
But how are histone acetylation and DNA methylation related to chromatin remodeling and gene expression? Nan et al. (32) and Jones et al. (33) demonstrated that gene silencing in eukaryotes involves the cooperation of DNA (cytosine) methylation and chromatin acetylation (Figure 3). In a series of follow-up studies (34–36), histone deacetylases were found to cooperate with histone methylases, establishing a “histone code” that resulted in self-propagating heterochromation assembly. Nakayama et al. (36) showed evidence that a conserved lysine residue, lysine 9 of histone 3 (H3K9), is preferentially methylated in heterochromatic regions of yeast and predicted that a similar mechanism might be responsible for higher-order chromatin assembly in humans.
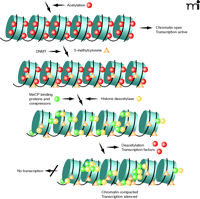
Furthermore, histone acetylation was associated with an open conformation of chromatin (euchromatin) allowing for gene transcription, whereas histone deacetylation permitted chromatin to assume and maintain a closed, non-transcribed state (heterochromatin). Bestor (37) explained that deacetylation of lysine ɛ-amino groups might allow greater interaction between positively charged N-terminal histone tails and the negatively charged DNA phosphate backbone. Methylation of CpG islands was important in gene silencing in such processes as X-inactivation, imprinting, and, possibly, in cancer (31). Aided by the genomics tools of molecular biology adapted for epigenetic analyses, several groups of investigators learned how the patterns of methylation were generated, read, maintained, and in most cases, faithfully transmitted from one generation to the next. Finally, a recent study has demonstrated that the human genome contains nearly 28 million CpG sites of which about 60% of the cytosines are methylated (38).
Histone Modifications
Alterations of chromatin structure are known to influence the cellular phenotype by recruiting activating or repressing chromatin protein complexes, resulting in an additional layer of complexity to epigenetic gene regulation (5). A striking feature of the core histones, particularly their tails, is the diverse array of posttranslational covalently modified amino-acid residues they possess (39, 40). These residues protrude from the nucleosome core and are accessible to enzymes that attach or remove these modifications. At least eight modifications of histone tail residues are known, although we have gleaned the most information about acetylation, methylation, and phosphorylation (39, 41, 42). Because distinct histone modifications correlate with specific transcriptional states, the “histone code” hypothesis (mentioned above) was proposed that these modifications dictate specific down stream events (41). For example, acetylation, methylation, phosphorylation, and ubiquinylation are involved in activation, whereas deacetylation, methylation, ubiquinylation, sumoylation, deimination, and proline isomerization are involved in repression. Under different conditions, however, it is possible that any given modification might act to activate or repress transcription.
Aberrant Epigenetic Patterns in Cancer
For more than fifty years, beginning with the discovery of the DNA double helix, the study of human disease has focused primarily on genetic mechanisms, but there is also considerable evidence that disruption of epigenetic pathways and networks are involved in the origin of major diseases. Thus far, cancer has been the main disease targeted by epigenetics research, but epigenetic mechanisms have been identified as causative of neurological and autoimmune disorders, and more recently have been implicated in the pathogenesis of cardiovascular disorders, metabolic diseases, obesity, and other nutritional disorders (23, 43), and in children born of assisted reproductive procedures (8, 44, 45).
During the last three decades, researchers have performed a broad range of phenomenological and mechanistic studies on the role of epigenetics in human carcinogenesis. Their findings have established that aberrant gene function and altered patterns of gene expression are key features of cancer and that epigenetic changes overwhelmingly involve DNA methylation (including genomic imprinting) and histone modifications.
The types of aberration observed in cancer most commonly include hypermethylation within gene promoter regions (46) and deacetylation with or without methylation of histone proteins. These abnormalities may act alone or together to alter the functions or expression of cellular component and they may occur at any stage in the development and progression of cancer.
Aberrant DNA Methylation in Cancer
Aberrant patterns in DNA hypomethylation and hypermethylation provided the first hints of epigenetic dysregulation in human cancer. Hypomethylation of individual genes and globally were first reported in cancer cells (47, 48). Typically, they exhibit hypomethylation of intergenic regions that usually contain the majority of the 5-methylcytosine content. As a consequence, transposable elements may be activated and contribute to genomic instability and chromosomal rearrangements, both of which may lead to further cancer-related events. Hypermethylation of promoters of genes involving important cellular pathways is another prominent feature of many major human tumor types (49, 50) and is a key event in carcinogenesis resulting from transcriptional silencing of tumor suppressor genes (50, 51). In addition, hypermethylation is often accompanied by global hypomethylation, an event that could affect cancer cells to a greater extent than coding region deletions or mutations, which are relatively rare.
Many studies and reviews have focused on the hypermethylation of genes and pathways, and processes or regions of functional importance (50–52). For example, an extensive study by Esteller and coworkers (50) analyzed twelve genes from over 600 primary tumor samples, representing fifteen major tumor types. They demonstrated promoter hypermethylation as well as simultaneous inactivation of several pathways by aberrant methylation in all fifteen tumor types. A more recent compilation by Cheung et al. (53) included cell-cycle genes plus genes that regulate apoptosis, detoxification, hormone response, Ras signaling and Wnt signaling, in addition to tumor suppression, DNA repair, and metastasis (50). Cheung et al. also described the role of hypomethylation of repetitive sequences such as Line 1, Alu, and transposable elements in promoting genome instability in various cancers (53).
The development of genome-wide methylation technologies has expanded understanding of DNA methylation patterns in normal cancerous cells (54). These assays have demonstrated that the genomic repetitive portion of normal cells is heavily methylated and most CpG islands are unmethylated, whereas cancer cells tend to exhibit widespread loss of intergenic DNA methylation with gain of methylation at many gene-associated CpG islands. They have also generated new information about DNA methylation patterns. For example, within individual tumors, 1–10% of CpG islands are aberrantly hypomethylated, and these normally methylated CpG islands become transcriptionally active. Still further, promoter-associated CpG islands are not the only islands affected by aberrant DNA methylation, as some CpG islands located within 3′ ends of genes and in intergenic regions exhibit hypermethylation in cancer cells, but whether, and to what extent, expression is affected in the non-promoter regions is unclear. Analysis of several genes with methylated 3′ CpG islands showed increased expression, suggesting a new function for DNA methylation in this location. Taken together, these alterations in methylations may have unanticipated effects on gene expression and cellular function than previously believed in cancer.
In another genome-wide study to determine the location of human colon-cancer related differential DNA methylation, Irrizary and colleagues (55) found that most alterations are not located in promoters or in CpG islands but rather are in sequences up to 2-kb distant, which they termed “CpG island shores.” Methylation at these “shores” is highly conserved and can define discriminating tissue types, regardless of their species of origin. They concluded that methylation changes in cancer are at sites that vary normally in tissue differentiation, and that epigenetic alterations affecting tissue-specific differentiation is the predominant mechanism by which epigenetic changes cause cancer.
Histone Modifications in Cancer
Epigenetic mechanisms implicated in aberrant chromatin packaging and remodeling, such as those that control transcription of genes involved in cell differentiation, proliferation, and survival, are often targets for deregulation in the development of cancer. The proteins responsible for the alterations that characterize the cancer epigenome include the enzymes that catalyze DNA methylation, the proteins that bind methylated DNA at promoters and contribute to silencing, and the chromatin modifer enzymes that catalyze various modifications of chromatin, particularly histone acetylation, deacetylation, methylation, and demethylation. Deregulation of these epigenetic modifiers has been characterized in many malignancies and plays a crucial role in certain other disorders (8, 23, 43). Specific examples of these protein modifiers are tabulated in reviews (56, 57). Extensions of this work, now widely accepted, have shown DNA cytosine methylation and histone modifications are intimately linked to nucleosome remodeling in cancer cells and that the interplay between all three of these mechanisms (i.e., cytosine methylation, histone modifications, and nucleosome remodeling) that result in permanent silencing of cancer-relevant genes may be deregulated in cancer (58).
Significant progress has been achieved in identifying aberrant histone modifications of chromatin during development and progression of carcinogenesis, a major function of which is to rearrange the chromatin environment in a fashion that is either permissive or repressive of gene transcription. Fraga and coworkers (59) first demonstrated global loss of monoacetylation at H4-Lys16 (H4K16ac) as well as loss of trimethylation of H4-Lys20 (H4K20me3) in a comprehensive study of a panel of normal tissues, cancer cell lines, and primary tumors. These changes appeared early and accumulated during the tumorigenic process. They were also associated with hypomethylation of DNA repetitive sequences of cancer cells. Fraga et al. interpreted the global loss of acetylation and trimethylation at histone 4 as hallmarks of human cancer cells.
Other investigators have shown that cancer cells exhibit a global decrease in H4K20me2/3, H3K9me2, and H4 acetylation, particularly at H4K16 (54). The loss of H4K16ac and H4K20me2/3 arises from the repetitive fraction of the genome and these changes occur in premalignant lesions and increase during tumor progression. Loss of DNA methylation (at H3K9me2 and H4K20me3) results in global dysregulation of transcriptional repression in cancer cells and may promote tumorigenesis through derepression of exogenous repetitive elements such as transposons, or miRNA, impaired DNA damage response, and chromosomal instability. In normal cells, an open chromatin structure marked by hyperacetylation of histones H3 and H4 and di- and trimethylation of histone H3 at lysine 4 (H3K4me2/3) constitutes a permissive region for transcription, whereas repressed regions exhibit a compact chromatin structure that lacks H3/H4 acetylation and H3K4 methylation, and is enriched instead in repressive modifications, di- and trimethylation of H3K9 (H3K9me2/3), trimethylation of H3K27 (H3K27me3), and trimethylation of H4K20 (H4K20me3).
The Challenge of Epigenetics to Personalized Medicine
Human cancer represents a group of heterogeneous disorders that is driven by combinations of genetic and epigenetic abnormalities. With regard to epigenetics, these disorders are characterized by aberrant cell- and tissue-specific patterns of DNA methylation and histone modification of chromatin that are reversible, interacting processes associated with inappropriate gene expression. Encouraged by the possibility that drug treatment can prevent or reverse the disease phenotype, researchers thought these aberrant patterns might be useful therapeutic targets in treatment of these diseases.
Early Attempts to Treat Epigenetic Disease
Numerous preclinical and clinical trials have sought to treat various hemoglobinopathies, myelodysplastic, and leukemic syndromes with demethylating agents, and the fragile X syndrome with demethylating agents (42, 60–64), histone deacetylating (HDAC) agents, or combined manipulation of cytosine methylation and histone acetylation. Agents used in these trials included older (5-azacytidine, 2-deoxy-5-azacytidine, or Decitabine®) and newer demethylating agents (MG98, an antisense oligonucleotide that decreases expression of DNMT1), and older (sodium butyrate, sodium phenyl butyrate) and newer HDAC inhibitors [trichostatin, suberoylanilide hydroxamic acid (SAHA) (65), and depsipeptide] (62, 63).
The emergence of epigenetic targets is an effective and valuable approach to cancer prevention and chemotherapy (66). These drugs can be used therapeutically, singly or as part of a combination with other therapeutic modalities such as chemotherapy, immunotherapy, or radiotherapy. Various HDAC inhibitors seem to enhance the tumor response to ionizing radiation. Thus, utilizing lesser but still therapeutically useful amounts of radiation may protect normal tissues from damage. Additionally, combinations of demethylating agents and HDAC inhibitors are also being studied for beneficial polypharmacy. Furthermore, the reactivation of tumor suppressor genes and DNA repair genes by epigenetic drugs would result in increases in chemosensitive cells. Epigenetic drugs could also help to alleviate resistance to other drugs by reactivating DNA-repair genes, such as MutL homolog 1(MLH1) or O-6-methylguanine-DNA methyl transferase (MGMT), thereby increasing the efficacy of existing therapies.
But at the same time, the risks of such therapy appear to be largely unknown. First, it is clear that hypomethylation is observed in malignant cells in vivo at doses of demethylating agents that overlap with doses that yield clinical responses. Second, multiple genes may be methylated, or undergo histone modification in epigenetic diseases, and because the therapeutic drugs listed above lack target specificity, there is the possibility of hitting many targets with one drug. The evidence for decitabine, for instance, favors DNMT inhibition as the central event in demethylation, but other possible events might include reactivation of tumor suppressor genes and activation of retrotransposons through hypomethylation. Furthermore, the extent to which other downstream events such as apoptosis and differentiation might occur is unclear. Finally, because methylation increases with age, inappropriate methylation might contribute to senescence and the development of age-related chronic diseases, including cancer, in the elderly (29–31).
In 2004, 5-azacytidine was the first agent to receive FDA approval for the treatment of several myelodysplastic syndromes. In 2006, the demethylating agent Decitabine® (5-2′-deoxyazacytidine) received FDA approval for treatment of myelodysplastic syndrome, and the HDAC inhibitor Zolinza® [SAHA, vorinostat (65)] received FDA approval for the treatment of cutaneous T cell lymphoma. Several other epigenetic drugs are capable of altering DNA methylation patterns or the modification of patterns of the tails of core histones. Those targeting methylation including 5-fluoro-2′-deoxycytidine (FCDR), epigalocatechin-3-gallate (EGCG), zebularine, procainamide, vorinostat, Psammaplin A, and antisense oligomers. Those targeting HDACs include phenylbutyric acid, SAHA, depsipeptide, and valproic acid. Some of these agents have been in clinical trials (30, 67).
Future Prospects that Might Occur Unexpectedly
DNA methylation and post-translational modifications of histones are important processes that regulate chromatin architecture, promoter activity, and, thus, cellular reprogramming. The mechanisms underlying the deposition of these marks, their propagation during cell replication, and alteration in their distribution during transformation are not fully understood and require further study.
Richly and colleagues (68) pointed out certain prospects that might occur unexpectedly, for example, in epigenetic reprogramming of cancer cells. During developmental reprogramming, the acquisition of a characteristic set of epigenetic marks is believed to specify terminally differentiated cell types. However, the mechanisms underlying the deposition of these marks, their propagation during cell replication, and their distribution during transformation are still unclear (68). They discussed ideas originally formulated by Conrad Waddington, who likened this developmental scenario to a marble ball falling through an “epigenetic landscape” (69), corresponding to a cascade of cellular divisions facing cells in the embryo that give rise to “intermediate” cells, that is, multipotent cells that have self-renewal capacity. More recent data indicate that the multipotent cells can, in turn, generate all terminally differentiated cell types of the mammalian body.
Following this line of reasoning, Richly et al. proposed a hypothetical example of an unscheduled expression of a single transcription factor that could cause the “reprogramming” of a mature cell into different types. Examples to support their proposal include the finding that expression of the muscle cell–specific transcription factor MyoD can convert fibroblasts into muscle cells, that the forced expression of CCAAT/enhancer-binding protein alpha (C/EBPα) can lead to conversion of B lymphocytes into macrophages, and that a cocktail of transcription factors (Myc, Oct4, Sox2, and Klf4) can convert a terminally differentiated cell to a pluripotent state. Richly et al also pointed out that the genetic lesions responsible for the large number of acute myeloid leukemias often suggests that an altered expression of transcription factors, including the formation of chimeric proteins (70), might be involved in the initiation of cancer. They raise the question of whether the unscheduled expression of such a transcription factor in hemopoietic cells could lead not only to an altered pattern of epigenetic marks but also to a reprogramming of hemopoietic progenitors into leukemic stem cells with the ability for self-renewal. The occurrence of such an event would obviously be of utmost concern.
Personalizing Epigenetic Therapy
Recently, the development of advanced sequencing protocols that combine chromatin immunoprecipitation (ChIP) with serial analysis of gene expression (SAGE), serial analysis of chromatin occupancy (SACO), and genome-wide mapping technique (GMAT) have permitted researchers to generate precise maps of epigenetic marks in different cell types (71, 72). Epigenomic maps in combination with the binding profiles of transcription factors have improved our understanding of regulation at promoters and of the organization of chromatin in health and disease.
Several inhibitors of chromatin modifying enzymes have been approved by the FDA. Because the technical capability to generate precise epigenomic maps is available, the Alliance for the Human Epigenome and Disease (AHEAD) project has recommended an international effort be undertaken to characterize the human epigenetic changes that accompany normal development, adult cell renewal and disease, and epigenetic variation that accompanies a healthy state with the goal of providing high-resolution reference epigenome maps. Substantial contributions have already been made toward understanding the importance of epigenetics to human disease and how to optimize its management, but we must learn much more about the proteins that are targeted by therapeutic agents, and the molecular interactions they perturb, before rationally designed drugs and individualized therapy becomes a reasonable goal outside researchers’ laboratories (29–31). As members of epigenome AHEAD project task force have noted (73), the epigenomic maps could have a major effect on understanding many human diseases and could lead to new means of disease control. These maps would be a valuable step toward realizing the personalization of epigenetics in medical practice.
- Copyright © 2010
Reference List

Wendell W. Weber, PhD, MD, is Professor Emeritus at the University of Michigan Medical School. E-mail wwweber{at}umich.edu; fax 734-763-4450.

