NOX Isoforms and Reactive Oxygen Species in Vascular Health
Abstract
Reactive oxygen species (ROS) are important mediators of cell growth, adhesion, differentiation, migration, senescence, and apoptosis. ROS play an important physiological role in regulating vascular tone and can also contribute to pathological mechanisms related to endothelial dysfunction, vascular reactivity, arterial remodeling, and vascular inflammation. The major source of ROS generated in the cardiovascular system is the NADPH oxidase (NOX) family of enzymes, of which seven members have been characterized. Although each NOX family member is typified by six transmembrane domains along with a cytoplasmic domain that binds NADPH and FAD, each isoform is distinguished by the specific catalytic subunit, interacting proteins, and subcellular localization. We review the current understanding of NOX signaling and regulatory mechanisms related to vascular health and disease.
Introduction
Reactive oxygen species (ROS) were originally considered solely as metabolic byproducts that, by virtue of their highly reactive free radical character, were deleterious to cell function and viability. But ROS are increasingly recognized as essential mediators of fundamental biological processes involved in development, hormone synthesis, extracellular matrix turnover, cellular senescence, apoptosis, anoikis, and oxygen sensing. This array of activities reflects the diverse roles of ROS as signaling molecules in cellular responses to growth factors, cytokines, hormones, and vasoactive agents (1). Within these contexts, ROS support the induction of host defense responses, the activation of transcription factors, and the regulation of kinases, phosphatases, and ion channels (2–4).
ROS are formed as intermediates in redox reactions (Box 1) that occur in all biological and organ systems, and the production of these reactive intermediates must accordingly be tightly regulated. ROS-generating enzymes include xanthine oxidoreductase, the mitochondrial respiratory enzymes, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. An additional set of enzyme activities act to delimit cellular ROS levels, comprising a highly regulated antioxidant system, which includes superoxide dismutase, catalase, and glutathione peroxidase, among others. Superoxide anion and hydrogen peroxide may be considered as important focal points amid the interplay of enzymatic redox reactions that generate and consume ROS. Superoxide ions, which are rapidly reduced to hydrogen peroxide by superoxide dismutase, are generally unable, owing to their negative charge, to cross cellular membranes. By contrast, H2O2 is relatively stable and readily diffuses within and between cells. The chemical properties that distinguish superoxide and hydrogen peroxide, and the fact that enzymatic turnover of these species occurs in distinct subcellular locations, likely contribute to the diversity of ROS-mediated signaling pathways in biological systems. Three isoforms of superoxide dismutase have been characterized in mammals: copper/zinc SOD (SOD1), mitochondrial SOD (SOD2), and extracellular SOD (EC-SOD, SOD3) (5–7). In the cardiovascular system, for example, the majority of superoxide dismutase activity is mediated EC-SOD.
Box 1. What a Difference an Electron Makes
The sequential univalent reduction of O2 is:
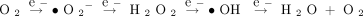
Of the ROS generated in vascular cells, superoxide anion (•O2−), and hydrogen peroxide (H2O2) appear to be particularly important. •O2 is short-lived because of its rapid reduction to H2O2 by superoxide dismutase (SOD) activities. The chemical properties that distinguish •O2− and H2O2, along with their genesis in distinct subcellular locations, contribute to the diversity of signaling mechanisms in which ROS are involved. ROS that induce oxidative stress include •O2−, H2O2, •OH, HOCl, and the reactive nitrogen species (RNS) nitric oxide (NO) and peroxynitrite (ONOO−).
Under conditions of oxidative stress, the balance between pro- and antioxidant activities becomes disrupted, and the bioavailability of ROS can rise to unhealthy levels, typically invoking pathways of inflammation, hypertrophy, proliferation, apoptosis, migration, fibrosis, angiogenesis, and rarefaction. These responses, in vascular cells, are related to vascular remodeling and endothelial dysfunction, which can set the stage for hypertension, atherosclerosis, obesity, diabetes, stroke, cardiac failure, and kidney disease (8–12). Oxidative stress further derails signaling in the cardiovascular system by impinging upon nitric oxide signaling. Specifically, oxidizing conditions undermine the dimeric stability of nitric oxide synthase (NOS), resulting in the monomeric, or uncoupled, form of the enzyme. Uncoupled NOS not only fails to produce NO but also exacerbates oxidative stress by generating superoxide anion. The bioavailability of NO is further depleted as it reacts with super-oxide anion to form peroxynitrite, and reactive nitrogen species thus represent challenges to cell function that are inseparable from those posed directly by ROS.
Research into the pathophysiolgical mechanisms that produce and depend on elevated ROS levels is a crucial part of ongoing efforts to combat cardiovascular disease. The challenge to elaborate the complex interplay of redox reactions and their sequelae in cardiovascular disease is formidable. One important thrust of attempts to understand and pharmacologically combat excessive ROS production focuses on the expression and regulation of the NADPH oxidase (NOX) isoforms and regulatory proteins that function to generate the majority of ROS in the cardiovascular system. We review these isoforms and discuss recent developments in our understanding of their regulatory mechanisms.
NADPH Oxidase: ROS Generation as a Full-time Profession
Of the many ROS-generating reactions that occur in cells throughout the body, the sole enzyme that produces ROS as its primary function is NOX; the enzyme has hence been termed a “professional” ROS producer (13). Originally designated as “phagocytic oxidase” (phox) activities involved in defense and innate immunity, the NOX family is now formally recognized as a category of trans-membrane proteins expressed by a wide variety of cell types. The NOX family includes the prototypical gp91phox (i.e., NOX2) as well as other isoforms that are important in vascular ROS production. NOX activity generally requires the association of a catalytic subunit (i.e., NOX) with other proteins. For example, NOX2 activity depends on at least five subunits: p47phox, p67phox, p40phox, p22phox, and the catalytic NOX2 subunit (14, 15) (Figure 1). Under basal conditions, p47phox, p67phox, and p40phox exist in the cytosol, whereas p22phox and NOX2 occur as a membrane-bound dimer (also known as cytochrome b558). Upon stimulation, p47phox is phosphorylated and forms a complex, with p67phox, that translocates to the membrane to establish oxidase activity (16); p40phox may stimulate or inhibit NOX function (17). NOX activation also requires participation of RAC and RAP1A GTPases (18).
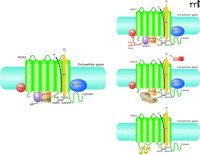
NOX2 is the prototypical structure, showing the six transmembrane domains and cytosolic termini. Regulation of the four NOX isoforms that are relevant to cardiovascular health is mediated by variations of NOX isoform structure as well as interactions with various protein factors. See Table 1 and text for details.
Each of the seven isoforms that belongs to the mammalian NOX family is encoded by its own respective gene. All NOX isoforms are transmembrane proteins that have conserved structural properties to subserve the reduction of molecular oxygen and produce superoxide anion in concert with the transport of electrons across the plasma membrane. Structurally, each NOX protein is defined by a cytoplasmic N terminus, six transmembrane domains (containing four conserved histidine residues that provide two heme-binding sites), and a long cytosolic C-terminal tail that contains the FAD- and NADPH-binding sites (Figure 1). NOX1, NOX2, NOX4, and NOX5 have been identified in vascular tissue (19, 20); NOX3, expressed in the inner ear, and DUOX1 and DUOX 2, found primarily in the thyroid gland, do not seem to be important in the vascular system.
NOX1
NOX1, found mainly in colon epithelial cells, is also expressed in other cell types, including endothelial and vascular smooth muscle cells (21, 22). NOX1 is similar to NOX2 in that its activity requires p22phox, p47phox [or the analogous NOXO1 (NOX organizer 1)], p67phox [or the analogous NOXA1 (NOX activator 1)], and RAC1 (23, 24) (Figure 1). Additional mechanisms of NOX1 regulation have been identified. TKS4 and TKS5, for example, along with NOXO1, have been reported to function as p47phox; NOXA1 appears to prevent NOX hyperactivity by engaging kinase pathways that inhibit interaction between NOX1 and RAC (25). TKS5 functions to facilitate ROS production necessary for invadopodia formation (26). NOX1 can constitutively produce low levels of superoxide anion, but these levels increase in a stimulus-dependent manner, involving complex interactions made between NOXO1, NOXA1, and membrane-bound p22phox (23). Significantly, NOXA1 may regulate NOX1 through PKC-, MAPK-, and PKA-dependent mechanisms that inhibit the interactions between NOX1 and RAC1 (27). NOX1 localizes with p22phox in caveolae and is expressed at low levels under physiological conditions. It is also regulated by protein disulfide isomerase (PDI), the redox chaperone protein, in vascular smooth muscle cells (28). NOX1 has been implicated in vascular smooth muscle cell migration, proliferation, and extracellular matrix production, effects mediated by cofilin (29).
In cultured endothelial and vascular smooth muscle cells, NOX1 is upregulated by shear stress, angiotensin II (AngII), aldosterone, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF) (30–34). AngII-regulated induction of NOX1 may be mediated through mitochondrial interaction, possibly reflecting a Ca2+-dependent mechanism (35). NOX1 is induced in the vasculature in models of hypertension, atherosclerosis, diabetes, and hypercholesterolemia (36). Studies of transgenic mice suggest a possible role for NOX1 in acute, but not chronic, forms of AngII-dependent hypertension (11, 37–39) and in atherosclerosis, restenosis post injury, endothelial dysfunction, and stroke (36). Despite the extensive experimental data implicating NOX1 in vascular disease, there is a paucity of clinical data, although expression of NOX1 and NOXA1 is increased in human atherosclerotic vessels (40).
NOX2
NOX2 is the catalytic subunit of the respiratory burst oxidase in phagocytes, but it is also expressed in vascular, cardiac, renal, and neural cells (30, 41–44). Full activation of human NOX2, a highly glycosylated protein, requires p22phox, p47phox, p67phox, and RAC (Figure 1). In neutrophils, NOX2 localizes to intracellular and plasma membranes; in vascular smooth muscle and endothelial cells, it also localizes with the cytoskeleton, lipid rafts/caveolae, and perinuclear compartment (45). The NOX2 gene, located on the X chromosome, is highly regulated by AngII and stretch.
The physiological functions of NOX2 in signal transduction or normal cell function in the vasculature remain unclear, but pathological increases in NOX2 contribute to oxidative injury and vascular damage, including angiogenesis, endothelial dysfunction, vascular inflammation, and structural remodeling. Vascular NOX2, derived from resident macrophages or vascular cells, is upregulated in experimental hypertension (46–48), atherosclerosis, ischemia-reperfusion injury, and neointimal formation (36). Although NOX2 has been shown to be important in models of AngII-infused hypertension, it does not seem to be critical in models of chronic AngII-dependent hypertension, given that transgenic Nox2−/− mice that overexpress human renin (with consequently increased AngII production) manifest AngII-dependent hypertension and cardiac hypertrophy (39, 49). NOX2 is also implicated in stroke; Nox2-deficient mice exhibit significantly reduced cerebral infarct size compared to wild-type controls (50). In humans, NOX2 has been shown to play a role in endothelial function, as patients who have an NOX2 mutation and concomitant chronic granulomatous disease exhibit a significant increase in forearm-mediated vasodilation with increased NO bioavailability (51, 52).
NOX4
NOX4, of which four splice variants have been identified (NOX4B, NOXC, NOX4D, and NOX4E), occurs in vascular cells, fibroblasts, and osteoclasts and is abundantly expressed in the kidney (53–56). In vascular smooth muscle cells, NOX4 colocalizes with p22phox and vinculin in focal adhesions and has been implicated in cell migration, proliferation, tube formation, angiogenesis, and cell differentiation (57, 58). NOX4 has been identified in the endoplasmic reticulum, mitochondria, and nucleus of vascular cells (58, 59) and may manifest an isoform-specific distribution. NOX4 activation does not seem to require p47phox, p67phox, p40phox, or RAC, although two NOX4-binding proteins (i.e., NOXR1 and Poldip2) have recently been shown to be important (60) (Figure 1).
Unlike NOX1 and NOX2, NOX4 is constitutively active, and of the ROS that it generates, H2O2 appears to predominate over superoxide anion (61), perhaps reflecting NOX isoform–specific roles in cell signaling. NOX4 constitutively contributes to basal ROS production, but ROS generation by NOX4 is increased in response to AngII, glucose, TNFα, and growth factors (62). Although the precise mechanisms are unclear, NOX4 has been implicated in hypertension, atherosclerosis, cardiovascular and renal complications of diabetes, and in the remodeling of pulmonary arteries during hypoxia-induced pulmonary hypertension (62–65). NOX4 has also been implicated in cellular senescence and aging (66–68) as well as in the insulin-mediated differentiation of adipocytes (69). Recent studies suggest that NOX4 may also have protective effects. Specifically, Nox4-null mice develop exaggerated contractile function, hypertrophy, and cardiac dilatation in response to chronic overload, whereas the cardiomyocyte-targeted expression of NOX4 protects transgenic mice from these effects (70).
NOX5
NOX5 is the most recently identified of the NOX isoforms, and its features are highly distinctive. NOX5 is regulated in a Ca2+-sensitive manner and is found in testes, spleen, and lymphoid tissue as well as in kidney and vascular cells (71, 72). The rodent genome does not encode a NOX5-expressing gene (73). Unlike the other vascular isoforms, NOX5 possesses an amino-terminal calmodulin-like domain with four binding sites for Ca2+. NOX5 is also unique in that its activation is independent of p22phox or other subunits (Figure 1). The biological significance of vascular NOX5 is unknown, although it has been implicated in endothelial cell proliferation, angiogenesis, and migration; it also appears to play a role in the PDGF-induced proliferation of vascular smooth muscle cells and in oxidative damage in atherosclerosis (74, 75). Vascular NOX5 is activated by thrombin, PDGF, AngII, and endothelin 1 (53, 74–76); its expression is regulated by the calcium-sensitive transcription factor CREB (77).
Distribution and Regulation of NOX Isoforms in the Vascular System
Endothelial cells, vascular smooth muscle cells, and adventitial fibroblasts express multiple NOX isoforms (36, 53, 78, 79). Significantly, mRNA expression profiles of NOX isoforms may not necessarily correspond to protein expression and activity profiles, likely reflecting the regulation of NOX activities through association with other proteins. In vascular disease, macrophages and leukocytes, which are rich in NOX2, take up residence in the vasculature and contribute to ROS production. Endothelial cells express NOX2, NOX4, and the regulatory proteins p22phox, p47phox, and p67phox (80). NOX2 and NOX4 are the major sources of ROS in endothelial cells under basal conditions; in pathological states, NOX1, NOX4, and NOX5 may be upregulated (81). NOX2, NOX4, and NOX5 localize mainly in the perinuclear area associated with the endoplasmic reticulum, although NOX2 is also found in the plasma membrane within cholesterol-enriched domains that may serve as signaling platforms for ROS generation in vascular disease (82). The major source of ROS in vascular smooth muscle cells arises from NOX2 (in human resistance arteries), NOX4, and NOX5. NOX1, expressed at low levels in basal states, is upregulated in disease (78, 79). Adventitial fibroblasts possess NOX2 and NOX4, and perivascular adipose tissue expresses NOX 4 (83, 84).
Regulatory Mechanisms of NOX Activity
The regulation of NOX activity is complex, involving the association of many protein subunits that may provide functionality within multiple signaling pathways (Table 1). All NOX isoforms, except NOX5, appear to have an obligatory need for p22phox (74, 85, 86). Whereas NOX2 requires p47phox and p67phox for its activity, NOX1 may interact with NOXO1 and NOXA1 (87, 88). Oxidase activation involves RAC translocation, phosphorylation and translocation of p47phox (possibly in association with p67phox), and p47phox association with the NOX2–p22phox dimer. NOX2 and NOX4 are constitutively active in intact vessels. Induction of NOX expression occurs in response to physical stimuli (e.g., shear stress and pressure), growth factors (e.g., PDGF, EGF, and TGFβ), cytokines (e.g., TNFα, IL-1, and PAF), mechanical forces (e.g., cyclic stretch and laminar and oscillatory shear stress), metabolic factors [e.g., hyperglycemia, hyperinsulinemia, free fatty acids, and advanced glycation end products (AGEs)], GPCR agonists (e.g., serotonin, thrombin, bradykinin, endothelin, and AngII), and hypoxia/hyperoxia (88–91). Transcriptional regulation of the NOX genes involves nuclear factor-κB (NF-κB), activator protein 1 and Nrf2, as well as changes in intracellular redox status (65, 92, 93). NOX activity has recently been shown to be regulated by CLCN3, Poldip2, and PDI. Poldip2 associates with p22phox to activate NOX4, leading to regulation of focal adhesion turnover and vascular smooth muscle cell migration, thus linking ROS production to cytoskeletal reorganization (60). AngII, an important and potent regulator of vascular NOX, acts through AT1 receptors to stimulate signaling pathways involving SRC, RAS, PKC, PLD, and PLA2 (65, 93–96).
NADPH Oxidases: Primary Producers of Reactive Oxygen Species in Vasculature
Conclusions
Extensive data confirm the importance of NOX-derived ROS in the physiological control of vascular function—specifically, in the regulation of endothelial function and vascular tone via redox-sensitive signaling pathways. The dysregulated turnover of NOX-generated ROS is a feature of oxidative stress and can result in cardiovascular, renal, and neural damage. ROS play diverse intracellular signaling roles, and the diversity and specificity of these roles depend on the tissue and subcellular localization and activation of the various NOX isoforms. NOX activity is regulated at the transcriptional and translational levels, involving many factors that may themselves be redox-sensitive. Although there is strong experimental evidence indicating a role for NOX-derived ROS in vascular disease, the clinical data are less conclusive. Future studies of NOX family members and their roles in ROS production and redox signaling in the vascular system will likely advance our understanding of oxidative stress and provide clinical opportunities for developing NOX-targeted therapies in human diseases.
Acknowledgments
RMT is supported by funds from the Canadian Institutes of Health Research (CIHR) [Grants 44018 and 57886] and by an award of the Canada Research Chair/Canadian Foundation for Innovation. DB and MS are supported by fellowships from the Kidney Research Scientist Core Education and National Training (KRESCENT) program, and ACM is supported through a fellowship from the CIHR. AMB was supported through a Beca de Posgrado from Fundación CajaMadrid.
Footnotes
-
Author contributions
Wrote or contributed to the writing of the manuscript: Touyz, Briones, Sedeek, Burger, and Montezano.
- Copyright © 2011
References
Rhian M. Touyz, MD, PhD (top left), is the Canada Research Chair in Hypertension and Professor of Medicine at the Kidney Research Centre, Ottawa Hospital Research Institute (OHRI), University of Ottawa. Ana M. Briones, PhD (center, left), is a visiting professor from the University of Madrid. Mona Sedeek, MBBCh, PhD (bottom, left), Dylan Burger, PhD (top, right), and Augusto C. Montezano, PhD (lower right), are research fellows in the Touyz laboratory at the Kidney Research Center, OHRI, University of Ottawa. Address correspondence to RMT. E-mail rtouyz{at}uottawa.ca; fax 613-562-5487.






