Marijuana-based Drugs: Innovative Therapeutics or Designer Drugs of Abuse?
- 1 Department of Pharmacology and Toxicology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR 72205
- 2 Department of Pediatrics, University of Arkansas for Medical Sciences, Department of Clinical Pharmacology and Toxicology, Arkansas Children’s Hospital, Little Rock, AR 72202
- 3 Arkansas Department of Health, Public Health Laboratory, Little Rock, AR 72205
Abstract
The principal psychoactive component of marijuana, Δ9-tetrahydrocannabinol (THC), activates CB1 cannabinoid receptors (CB1Rs). Unfortunately, pharmacological research into the design of effective THC analogs has been hampered by psychiatric side effects. THC-based drug design of a less academic nature, however, has led to the marketing of “synthetic marijuana,” labeled as K2 or “Spice,” among other terms, which elicits psychotropic actions via CB1R activation. Because of structural dissimilarity to THC, the active ingredients of K2/Spice preparations are widely unregulated. The K2/Spice “phenomenon” provides a context for considering whether marijuana-based drugs will truly provide innovative therapeutics or merely perpetuate drug abuse.
Marijuana has been used recreationally and medicinally for centuries. The principle psychoactive component, Δ9-tetrahydrocannabinol (Δ9-THC), activates CB1 cannabinoid receptors (CB1Rs). CB1R agonists and antagonists could potentially treat a wide variety of diseases; unfortunately, therapeutic doses produce unacceptable psychiatric effects. “K2” or “Spice” (K2/Spice), an emerging drug of abuse, exhibits psychotropic actions via CB1R activation. Because of structural dissimilarity to Δ9-THC, these drugs are widely unregulated and touted as “legal” marijuana. This review summarizes current and future therapeutic uses of CB1R ligands and provides a historical perspective of the K2/Spice “phenomenon” so the reader can decide if marijuana-based drugs will truly provide innovative therapeutics or instead perpetuate drug abuse.
Introduction
Although controversial, marijuana has been used medicinally for centuries (1) and several clinically available products of either Δ9-tetrahydrocannabinol (Δ9-THC, the principal psychoactive component of marijuana) or structurally related derivatives are available for safe and efficacious treatment for several diseases when administered appropriately and under proper medical supervision (2). Unfortunately, Δ9-THC-based drugs produce both therapeutic and undesirable psychotropic actions by activating CB1 cannabinoid receptors (CB1Rs) in the central nervous system (CNS).
K2, also called “Spice” (K2/Spice), is a rapidly emerging drug of abuse that is touted as “legal marijuana” [reviewed in (3)]. Although structurally distinct from Δ9-THC, the synthetic compounds in K2 products are derivatives of the well-characterized aminoalkylindole (AAI) class of ligands that also bind and activate CB1Rs. Because AAIs are structurally dissimilar to Δ9-THC, government regulation of K2 products in many states is inconsistent or even lacking, leading to widespread use and heavy marketing to teens and first-time drug users. In marked contrast to the relatively “safe” and established consequences of marijuana use, many symptoms associated with K2/Spice use are distinct from those of marijuana and may be potentially life threatening. Therefore, users experimenting with K2 products may, in fact, be exposed to a drug that is extremely variable in both composition and potency, and may develop serious adverse effects.
This review will provide a brief introduction to basic cannabinoid pharmacology, followed by a summary of current and future therapeutic uses of marijuana-based CB1R agonists and antagonists. Finally, a historical perspective of the K2/Spice “phenomenon” from inception to present day use of novel synthetic cannabinoids as designer drugs of abuse will be presented. The purpose of this review is to provide adequate information to allow readers to decide if the future development of marijuana-based drugs will truly provide innovative therapeutics or instead merely perpetuate the use of novel designer drugs of abuse. Although much evidence suggests that drugs acting at CB2Rs might also be developed as very efficacious agents to treat inflammation and other disorders [reviewed in (4)], this review will focus only on the therapeutic potential for drugs acting at CB1Rs.
Historical Perspective of Marijuana and Cannabinoids
Medicinal and Recreational Uses
Since the earliest known use of Cannabis sativa fibers found in China dating from around 4000 BCE, cultures throughout the world have been using cannabis-derived products (Figure 1) (1). In many ancient cultures (e.g., Chinese, Indian, and Tibetan), the seeds and fruit were used to treat a variety of ailments including gastrointestinal disturbances, seizures, malaria, pain of childbirth, snake bites, and many more (5). Aside from medicinal usage, the psychoactive properties of cannabis were also realized, and its use in religious ceremonies, such as those conducted by Tantric Buddhists, Hindus, and ancient shamans (5), became common. In addition to medicinal and religious uses, cannabis has been widely employed as a recreational drug for centuries (1).
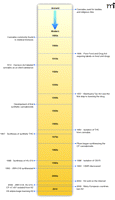
Timeline of ancient cannabis use to modern day banning of K2 products.
By the end of the nineteenth century, use of cannabis for medicinal purposes had grown and was supported by early scientific articles. As such, drug companies began marketing cannabis extracts as medicinal tinctures. In the early twentieth century, passage of the Pure Food and Drug Act of 1906 in the United States improved the safety of food and medicinal products by requiring labels to note dangerous contents, including cannabis (6). After additionally passing the Harrison Act of 1914, which grouped cannabis together with other illicit drugs such as cocaine and heroin, the marketing and use of cannabis-derived products decreased significantly. Reduced commercial interest in cannabis also corresponded with the development of more efficacious, safer drugs and with political pressure exerted by drug companies (1, 6). Subsequently, recreational use of cannabis products (such as marijuana) grew, and fear of the increasing marijuana abuse resulted in passage of the Marihuana Tax Act of 1937. Soon after implementation of this act, individual states subsequently outlawed the use, possession, or distribution of marijuana and other cannabis products (1, 6). Although illegal, recreational use soared during the 1960s and 1970s and continues to be high today, where approximately 50% of the population of the United States is reported to have tried cannabis at least once (7).
Isolation of Δ9-Tetrahydrocannabinol (Δ9-THC), the Psychoactive Component of Marijuana
Prior to the 1960s, few modern scientific studies had been published regarding cannabis. In 1964, Gaoni and Mechoulam first identified the principle psychoactive compound, Δ9-tetrahydrocannabinol (THC) (8). This renewed interest in the cannabinoid field and led to the isolation and synthesis of many additional cannabinoids and eventually the discovery of the endogenously occurring cannabinoids, the endocannabinoids (see following and Figure 1). Until as recently as the late 1980s, most scientists believed the effects of cannabis were not receptor-mediated because of the highly lipophilic nature of cannabinoids, which allowed these compounds to easily diffuse into and across cell membranes (9). In 1988, the first cannabinoid receptor (now classified as CB1) was isolated (10) and subsequently cloned (11). Discovery of CB1Rs sparked renewed interest in cannabinoids as possible therapeutic candidates.
Discovery of CB1 and CB2 Receptors
CB1 receptors (CB1R) are constitutively active pertussis toxin–sensitive Gi/o-coupled G protein coupled receptors (GPCRs) (12). CB1Rs are located throughout the body, with the highest density expressed in the central nervous system (CNS). They are involved in the regulation of many physiological processes, including energy balance (13) as well as arterial tone and vasorelaxation (14). CB1Rs are the most abundant GPCRs in the CNS, expressed in densities similar to that of γ-amino butyric acid (GABA) and glutamate receptors. The greatest populations of CB1Rs are located in the hippocampus, basal ganglia, and cerebellum (15). Agonists for CB1Rs, such as Δ9-THC, produce psychoactive properties characteristic of cannabis use (16). However, because very few CB1Rs are located in the brain stem (which controls vital physiological functions), Δ9-THC and other CB1R agonists have relatively low toxicity except at extremely high doses (16).
In the early 1990s, a second cannabinoid receptor (CB2R) was molecularly cloned (17). Like the CB1R, CB2Rs are constitutively active pertussis toxin–sensitive Gi/o-coupled GPCRs (18). Unlike CB1Rs, CB2Rs are primarily localized to immune cells in the periphery, that is, outside of the CNS (18). The greatest concentrations of CB2R mRNA are present within the spleen, tonsils, and thymus (19); thus, CB2Rs are recognized to be involved in modulation of immune function (18). Although normally absent from the CNS, during neuroinflammatory conditions CB2Rs can be found within the brain and brain stem (20) where CB2Rs are expressed on microglia, the resident macrophages of the CNS (18). Selective CB2R agonists do not produce psychoactive effects but instead suppress immune function.
When an agonist such as Δ9-THC binds these CB1Rs and CB2Rs, adenylyl cyclase activity is inhibited and intracellular concentrations of cAMP are reduced (21) (Figure 2). In addition, activation of both CBR types results in phosphorylation and stimulation of the mitogen-activated protein kinases extracellular signal–regulated kinase 1(ERK1) and ERK2 (12, 14). CB1R, but not CB2R, agonists additionally inhibit distinct voltage-gated calcium channels and activate inwardly rectifying potassium channels (12, 14).
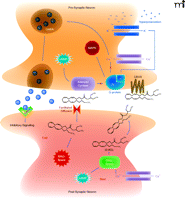
Endocannabinoids are produced on demand in post-synaptic neurons by stimuli that result in elevations of intracellular Ca2+ levels, activating synthetic enzymes such as diacylglycerol lipase (DAG-lipase). Synthesized endocannabinoids such as 2-arachidonyl glycerol (2-AG) then diffuse into the synaptic space, travel in a retrograde manner, and bind to CB1Rs on presynaptic neurons. Retrograde signaling produced by endocannabinoid binding to presynaptic CB1Rs results in inhibition of adenylyl cyclase and activation of mitogen-activated protein kinase (MAPK) activity. However, the inhibitory actions of cannabinoids on neurotransmission result primarily from hyperpolarization of presynaptic neurons resulting from inhibition of Ca2+ influx via voltage-gated Ca2+-channels and loss of intracellular K+ ions via opening of inwardly rectifying cannels. Hyperpolarized presynaptic neurons produce less exocytosis of neurotransmitter, such as γ–aminobutyric acid (GABA), resulting in decreased neurotransmission. Endocannabinoid actions are finally terminated by hydrolysis due to action of presynaptically located metabolizing enzymes such as monoacylglycerol lipase (MAG-lipase).
Cannabinoid Ligands Are Derived from Diverse Structural Groups
In general, cannabinoids are a diverse group of compounds containing natural, synthetic, and endogenous ligands that act selectively at one receptor or nonselectively at both cannabinoid receptors (12). Ligands that bind to CBRs are derived from four basic structural classes (Figure 3) [reviewed in (22)]. First, the classical cannabinoids (Figure 3A) consist of dibenzopy-ran derivatives (including Δ9-THC), which act as partial agonists at CB1R and CB2Rs. Also included in the classical group are other plant cannabinoids, such as the partial CBR agonist cannabinol, cannabidiol, (a natural cannabinoid devoid of psychoactive effects), and various synthetic cannabinoids such as HU-210 (a full agonist at both receptors) (23). Second, the nonclassical cannabinoid group (Figure 3B) lacks a pyran ring structure and includes cannabinoids such as CP-55,940, a potent agonist at CB1R and CB2R. Third, the aminoalkylindole group (Figure 3C) of cannabinoids includes WIN-55,212-2, which possesses slightly higher affinity for the CB2R than for CB1R. Lastly, the eicosanoid group of cannabinoids (Figure 3D) includes the endocannabinoids, their synthetic derivatives, and noladin ether, a putative endogenous cannabinoid that binds with highest affinity to CB1R. It is important to note that one of the major differences between Δ9-THC and almost all synthetic cannabinoids is that whereas the former is a weak partial agonist at CBRs, the latter group of compounds (including HU-210, CP-55,950, WIN-55,212-2, and many of their derivatives, Figure 3) are potent full CBR agonists (16).

Ligands that bind to CBRs are derived from four basic structural classes: (A) classical cannabinoids, (B) nonclassical cannabinoids, (C) aminoalkylindoles, and (D) eicosanoids.
Characterization of Endogenous Cannabinoids
While CB1Rs and CB2Rs were being discovered, the identification of the endocannabinoids was also reported. The first endocannabinoid discovered was anandamide (N-arachidonoylethanolamine or AEA), isolated from porcine brain (24). Soon after, two laboratories simultaneously isolated and reported the discovery of a second endocannabinoid, 2-arachidonyl glycerol (2-AG) (25, 26). AEA is a partial agonist at CB1Rs (27) and a partial agonist or antagonist at CB2Rs (28). In contrast, 2-AG demonstrates full agonist activity at both CB1Rs (29) and CB2Rs (30). Although both endocannabinoids are metabolites of arachidonic acid and synthesized “on demand” (31), 2-AG is found in much higher concentrations in the brain (29). AEA is synthesized from phosphatidylethanolamine via enzymatic conversion by transacylase and phospholipase D (31). Similarly, 2-AG is produced from phosphitidylinositol by action of the enzymes phospholipase G and diacylglycerol lipase (31). After synthesis, both endocannabinoids are immediately released into the intracellular space (31) and interact with not only classically recognized cannabinoid receptors, but also with less well characterized GPCRs (e.g., GPR55) and ion channels (e.g., vanilloid receptor-type1) (12). Cellular re-uptake of endocannabinoids is still not well characterized and controversial [reviewed in (32)]; however, once endocannabinoids are returned to the cytoplasm, AEA undergoes rapid hydrolysis, primarily by the microsomal enzyme fatty acid amid hydrolase (FAAH) (31), whereas 2-AG is inactivated predominantly by mono-glyceride lipase (MGLL) (31).
The involvement of the endocannabinoid system has been implicated in numerous disease states such as obesity, cardiovascular disease, and neurological disorders (33). The concentrations of endocannabinoids may change throughout the course of many diseases, and it has yet to be determined whether decreasing or increasing these concentrations may be advantageous in treating various diseases (34).
Behavioral Effects of CB1R Agonists
In addition to psychoactive effects that are elicited by CB1R activation in humans, acute administration of Δ9-THC or other CB1R agonists in rodents results in quantifiable actions, typically measured as a “tetrad” of behavioral effects composed of analgesia, catalepsy, hypothermia, and decreased locomotor activity (35). Administering Δ9-THC to humans also results in physical relaxation, changes in perception, mild euphoria, reduced motor coordination, and decreased information processing (36). Furthermore, Δ9-THC, AEA, and other CB1R agonists stimulate appetite even in satiated animals (37), indicating an involvement of the endocannabinoid system in regulating energy balance (13). Synthetic analogs of Δ9-THC such as nabilone (Cesamet®) (Figure 3) have been developed to stimulate appetite and reduce nausea in cancer and AIDS patients. In contrast, inverse CB1R agonists reduce eating and promote weight loss (38).
When Δ9-THC is administered chronically, tolerance develops to the acute effects owing, in part, to receptor down-regulation (39) and desensitization (40). Chronic and heavy use of cannabis has also been correlated with mild impairment of cognitive function, including decreased attention, verbal learning, and memory (41). It is unclear, however, whether these cognitive effects following chronic Δ9-THC exposure result from some type of adaptive process or instead arise from the acute actions of cannabis that are reversed upon cessation of drug exposure (36). Although only about 10% of chronic users develop dependence (36), chronic exposure to Δ9-THC nevertheless increases the likelihood of the individual to develop psychiatric disorders (42).
Δ9-THC also acts on CB2Rs. Because these receptors are primarily located on immune cells, CB2Rs are not believed to impact behavior or produce euphoria or the other psychoactive effects that are noted with CB1R agonist administration. Rather, the well-known immunosuppressive effects of Δ9-THC are most likely mediated via activation of these CB2Rs (43).
Therapeutic Promise of Cannabinoids
Therapeutic Versus Adverse Effects of CB1R Ligands in Drug Development
Great opportunity exists for the development of cannabinoid-based drugs for a wide range of therapeutic applications (44). However, only three prescription products containing the non-selective cannabinoid agonists nabilone (Cesamet®), dronabinol (Marinol®), and Δ9-THC/cannabidiol (Sativex®) are currently available clinically for use as antiemetics, appetite stimulants, and analgesics (Figure 3) [reviewed in (45)]. Animal studies suggest that future development of selective CB1R agonists may provide many additional drugs that will prove useful for treatment of epilepsy, inflammation, neurode-generation, cancer, anxiety, depression, and osteoporosis [reviewed in (46)]. CB1R antagonists–inverse agonists (such as rimonabant and taranabant) are also very effective for management of obesity in humans (47), and preclinical studies indicate that drugs in this class might be beneficial in treating drug abuse disorders, type-2 diabetes, liver fibrosis, certain types of inflammation, and psychosis [reviewed in (48)]. Unfortunately, in addition to the many potential beneficial uses, therapeutic doses of either CB1R agonists or antagonists–inverse agonists exert many undesirable psychological, physiological, and behavioral effects, greatly limiting their use. For example, in many patients CB1R agonists also produce euphoria, dysphoria, memory disturbances, tolerance, dependence, withdrawal, diminished psychomotor performance, and increased appetite (49). Several adverse effects, including nausea, depression, and suicidal ideation, are similarly associated with chronic blockade of central CB1Rs by antagonists (50). In fact, the prevalence and severity of these negative consequences led to the abrupt discontinuance of all ongoing clinical trials of CB1R antagonists–inverse agonists in Europe and the United States [reviewed in (51)].
Therefore, while a tremendous potential exists for future development of efficacious drugs targeting CB1Rs for a variety of disease states, it will be essential to take measures to circumvent associated adverse effects occurring at therapeutic doses so that successful clinical development may be realized. The following sections of this review will first briefly summarize the current therapeutic indications for use of CB1R agonists and antagonist-inverse agonists followed by a description of several potential avenues to minimize adverse, while maintaining beneficial, activity of these potentially valuable compounds. Finally, a brief listing of prospective future therapeutic uses for CB1R ligands, based on encouraging ongoing basic research findings, will be presented.
Current Licensed Therapeutic Uses of Δ9-THC and Other CB1R Agonists
Although cannabis as been used (and abused) for centuries, individual cannabinoid compounds were only made available for clinical use relatively recently [reviewed in (45)]. The first product, licensed in 1981, nabilone (Cesamet®), a synthetic derivative of Δ9-THC, is prescribed to reduce nausea and vomiting associated with cancer chemotherapy (52). In 1985, Δ9-THC, marketed as dronabinol (Marinol®), was approved as an antiemetic for use in similar situations (53), and in 1992 was additionally marketed to stimulate appetite in conditions associated with excessive weight loss such as that occurring in AIDS (54). The newest clinically available cannabinoid, Sativex®, is an equal mixture of Δ9-THC and a plant-derived cannabinoid lacking psychotropic effects known as cannabidiol (55). This combination product was approved in 2005 for management of neuropathic pain associated with multiple sclerosis and pain occurring in end-stage cancer patients (56). Interestingly, inclusion of the nonpsychoactive component cannabidiol in Sativex® appears to reduce many of the adverse effects, such as dysphoria and sedation, produced when synthetic cannabinoids such as Δ9-THC or nabilone are administered alone (57). Although Δ9-THC and nabilone are agonists at both CB1Rs and CB2Rs, the therapeutic effects for all marketed products thus far are thought to occur primarily in response to activation of CNS-located CB1Rs (45). The therapeutic applications approved for use of CB1R agonists described here have proven to be very valuable for use in a limited number of pathophysiological conditions. Further development of CB1R agonists, however, has been significantly hampered by not only adverse psychotropic effects but also by the social stigma often associated with their use.
Recent Clinical Trials for Therapeutic Use Of CB1R Inverse Agonists
Obesity is a major health problem in the United States where as many as 65% of adults are classified as overweight, 30% as obese, and 5% as morbidly obese (58). Although the toll of obesity on human mortality and morbidity is great (59), relatively few efficacious drugs (most with significant adverse effects) are available for treatment (60), and no new drugs have been successfully introduced into this market in over ten years (61). Many recent studies support a hypothesis that obesity is associated with an overactive endocannabinoid system resulting from enhanced levels of endogenously produced cannabinoids acting at CB1Rs in both the CNS and periphery (62). Activation of CB1Rs by endocannabinoids in the CNS stimulates appetite and controls the metabolic processes that mimic the metabolic syndrome (63), whereas stimulation of CB1Rs in peripheral tissues such as the liver, pancreas, adipose tissue, and skeletal muscle results in an overall slowing of metabolism and increase in visceral fat deposition in humans (64). As might be anticipated, blockade of excessive endocannabinoid signaling by use of CB1R antagonists–inverse agonists such as rimonabant (65) and taranabant (66) reduces weight and reverses many of the effects produced by an overactive endocannabinoid system occurring in obese animals and individuals. Furthermore, several large-scale clinical trials suggest that rimonabant also improves many other measures of the metabolic syndrome often occurring in conjunction with obesity. For example, the one-year RIO (Rimonabant In Obesity) trial demonstrated that daily rimonabant improves several obesity-associated cardiometabolic risk factors (67). The SERENADE (Study Evaluating Rimonabant Efficacy in Drug Naïve Diabetic Patients) trial further reported not only body weight reduction but also an improvement in lipid profiles and glycemic control in drug-naïve type 2 diabetics in response to six months of daily rimonabant treatment (68). Lastly, the STRADIVARIUS (Strategy to Reduce Atherosclerosis Development Involving Administration of Rimonabant—The Intravascular Ultrasound Study) trial demonstrated that rimonabant given daily for eighteen months normalized total atheroma volume in patients with atherosclerosis (69). Unfortunately, it also became evident from these same clinical trials that chronic blockade, inverse agonism, or both of central CB1Rs results in several significant adverse effects, including nausea, depression, and suicidal thoughts (66, 70). The severity of these side effects, in fact, led to discontinuance of all ongoing clinical trials of the CB1R antagonist–inverse agonist rimonabant in Europe (51), virtually assuring a lack of future FDA approval for use in the United States. Therefore, shortly after this decision was announced, all ongoing clinical trials and drug development programs directed toward discovery of novel CB1R antagonists–inverse agonists by several pharmaceutical companies in both the United States and Europe were terminated [reviewed in (47)]. Thus, it appears that either stimulation or blockade of CB1Rs located in the CNS by currently available drugs results in unacceptable side effects that, at present, hamper future development of this potentially valuable drug class.
Potential Mechanisms to Circumvent Adverse Effects Associated with Use of CB1R Ligands
Several avenues are being pursued to maximize the beneficial effects, while minimizing the negative consequences, of CB1R agonists and antagonists–inverse agonists with a purpose of enhancing efforts for future drug development (Table 1).
Strategies to Circumvent Adverse Effects of CB1R Ligands
Peripherally Restricted CB1R Ligands
Activation or antagonism of CB1Rs located in the CNS results in unacceptable adverse psychiatric-associated effects (51). However, stimulation of peripherally located CB1Rs is also effective at suppressing inflammation that leads to chronic pain states (71), and many of the positive metabolic benefits produced by CB1 antagonists–inverse agonists in obese animals are because of action at peripheral CB1 receptors (65). Therefore, development of selective CB1R ligands demonstrating relatively poor entry into the CNS may constitute a new class of therapeutic cannabinoids referred to as “peripherally-restricted CB1R ligands” [reviewed in (48)]. Drugs in this class would be predicted to demonstrate a markedly safer therapeutic profile than that observed for currently available CB1 agonists or antagonists. Significant progress has been made in this area and recent papers report the initial characterization of two peripherally restricted cannabinoid ligands URB447 (72) and SAB378 (73). URB447 is a nonselective CB1R antagonist and CB2R agonist that significantly reduces food intake and body weight in mice, and SAB378 is an agonist at both CB1Rs and CB2Rs shown to inhibit gastrointestinal motility.
CB1R Allosteric Modulators
Like many GPCRs, CB1Rs contain both orthosteric and allosteric binding sites (74). Endocannabinoids bind to orthosteric receptor sites, but binding of drugs to an allosteric site alters the affinity, efficacy, or both produced by orthosteric ligands (75). Therefore, drugs acting at these distinct sites are referred to as allosteric modulators (AMs) because they are able to “fine-tune” the actions of endogenously produced and synthetically administered or endogenously produced drugs acting concurrently at orthosteric sites. AMs can either increase [e.g., positive AMs or (PAMs)] or decrease [e.g., negative AMs of (NAMs)] the potency and efficacy of endocannabinoids; thus, these drugs might overcome the many undesirable effects observed by either direct CB1R activation or blockade in the CNS by currently available drugs. For example, in diseases where endocannabioid concentrations are enhanced (e.g., obesity), NAMs would selectively decrease CB1R signaling exclusively in tissues where elevated amounts of endocannabinoids are found. Similarly, in conditions where enhancement of CB1R signaling is beneficial (e.g., neuropathic pain), PAMs would exclusively increase endocannabinoid action at CB1Rs in a tissue-or neuron-specific manner. In both examples, AMs would provide an exquisite temporal and spatial modulation of CB1R signaling to limit off-target, systemic toxicity. In addition, homeostatic endocannabinoid concentrations in noninvolved tissues would likely be maintained. Lastly, owing to reduced evolutionary conservation of allosteric relative to orthosteric GPCR binding sites, AMs exhibit markedly greater receptor-subtype specificity when compared to orthosteric ligands (76). Consequently, use of CB1R AMs would produce a very selective enhancement of the action of locally released endocannabinoids only at CB1Rs. Although no high-affinity selective PAMs for CB1Rs have yet been discovered, different groups have reported identification of two structurally distinct classes of NAMs that selectively modulate CB1R activity at nanomolar concentrations (74, 77). Interestingly, while compounds derived from both structural classes of NAMs decrease the function produced by coadministered CB1R agonists, they (counter-intuitively) increase agonist binding affinity. Importantly, one of the CB1R NAMs identified (PSNCBAM-1) also produces hypophagia and body weight–reduction in an acute rat-feeding model that is similar to that observed previously for the direct acting CB1R orthosteric antagonist rimonabant (77). Furthermore, PSNCBAM-1 also acts as a CB1R allosteric antagonist to modulate neuronal excitability in mouse cerebellum (78), suggesting that it or other similar compounds may be useful for treatment of certain CNS disorders. Whether these compounds will exhibit less adverse psychiatric effects relative to currently available orthosteric CB1R ligands, as hoped, has yet to be determined.
Inhibition of Endocannabinoid Degradation
In many diseases, endocannabinoids are released in a tissue-specific manner in response to injury as a protective response (79). Endocannabinoids such as AEA and 2AG are quickly metabolized by the enzymes FAAH and MGLL, respectively; thus, the beneficial response observed in many cases is short-lived. Therefore, a pharmacological intervention strategy to avoid adverse effects of directly administered synthetic CB1R agonists is, instead, to enhance and maintain selectively the amounts of endocannabinoids in diseased tissues [reviewed in (32)]. In this case, activation of CB1Rs (and CB2Rs) is achieved indirectly by elevating endocannabinoid levels via blockade of their breakdown (80). Theoretically, CB1R activation would be localized to the tissues in which the elevated amounts of endocannabinoids occur, limiting adverse effects. One (of many) “proof of principle” study has demonstrated the therapeutic promise of such a strategy by demonstrating that blockade of degradation elevates the amount of endocannabinoids in local tissue, resulting in a reduction of inflammatory pain (81). However, a recent report also showed that elevated 2AG, but not AEA, levels owing to chronic blockade of their respective degrading enzymes results in functional antagonism of CB1R signaling (82). This finding suggests that approaches employed to activate indirectly CB1Rs may not prevent all adverse effects associated with administration of direct CB1R agonists. Furthermore, because all endocannabinoids identified to date activate both CB1Rs and CB2Rs nonselectively, these strategies would primarily be beneficial only in specific diseases in which concurrent activation of both CB1Rs and CB2Rs is desired. Several selective inhibitors of either FAAH and MGLL for in vivo use are commercially available, examples of which are URB-597 (an FAAH inhibitor) (83) and JZL-184 (an MGLL inhibitor) (84). Although less well characterized and often controversial, an alternative mechanism to increase extracellular levels of AEA is to block the cellular uptake of this endocannabinoid by use of putative anadamide transport inhibitors (85).
Use of Neutral CB1R Antagonists
CB1Rs, like many other GPCRs, exhibit constitutive activity or basal receptor signaling in the absence of activation by agonists (86). Antagonists, which simply occupy the receptor and block the binding of endogenous agonists, have no effect on constitutive GPCR activity and are referred to as “neutral antagonists.” In contrast, inverse agonists not only bind to CB1Rs but also preferentially reduce their constitutive activity, thus producing effects that are opposite to that observed for agonists (87). Importantly, all CB1R antagonists-inverse agonists examined in clinical trials to date actually act as inverse agonists, and not as neutral antagonists (88). Therefore, it is possible that the adverse effects observed for currently available CB1R inverse agonists occur, in part, because of their interference with constitutive or intrinsic cellular endocannabinoid signaling. Evidence from animal studies suggests that this might indeed be the case (89). Thus, development of neutral CB1R antagonists (that only alter ligand-dependent and not constitutive receptor signaling) might maximize the beneficial effects while minimizing the negative consequences of this potentially valuable class of drugs [reviewed in (48)].
Potential Future Therapeutic Uses for CB1R Ligands
The endocannabinoid system is ubiquitously expressed throughout the body and is responsible for the homeostatic control of many basic physiological processes (90). As might be expected, perturbation of this important system appears to contribute to (or result from) the pathophysiological mechanisms underlying a number of disease states (79). In disorders where abnormally low amounts of endocannabinoids and reduced CB1R signaling contribute to pathology or where CB1R activation is protective, the use of CB1R agonists appears to be beneficial. For example, drugs that enhance CB1R signaling are particularly useful in animal models of neurodegenerative diseases, neuropathic and other diverse types of pain, epilepsy, inflammation, gastrointestional disorders, cancer, anxiety, osteoporosis, and depression [reviewed in (46)]. On the other hand, disease states characterized by excessive endocannabinoid production and overactive CB1R signaling seem to respond positively to treatment with CB1R antagonists–inverse agonists. Therefore, drugs that reduce or block CB1R signaling are exceptionally effective in animal models of obesity, type-2 diabetes, drug abuse, psychosis, and chronic liver fibrosis [reviewed in (48)]. The key to successful development of efficacious agents derived from this valuable drug class will rely on future basic research to design highly selective CB1R ligands coupled with measures to limit associated adverse effects and to understand properly the distinct type of endocannabinoid dysregulation occurring in the specific diseases targeted for treatment.
Use of Novel Synthetic Cannabinoids as Designer Drugs of Abuse
Historical Perspectives of Δ9-THC Derivative Development
The history of the development of synthetic Δ9-THC and various Δ9-THC analogs provides insight as to why cannabinoids have recently begun emerging as new designer drugs worldwide (Figure 1). It is thought that Δ9-THC was first synthesized as a chemical intermediate to aid in the production of cannabinol, which at the time was thought to be the active constituent in C. sativa (91). In 1964, Δ9-THC was determined to be the pharmacologically active component of C. sativa, which led to additional synthesis efforts (8, 92–94). Synthetic Δ9-THC was produced in 1967 (91), and in the late 1960s the Levo (l)-isomer was identified as the biologically active form (8, 95–97). Early synthetic preparations contained both the Dextro (d)- and (l)-isomer, but improved synthetic and purification strategies provided means for isolating the biologically active isoform. In 1985, synthetic Δ9-THC was approved in the United States as an anti-emetic drug and marketed as Marinol® (dronabinol).
Classical Cannabinoids
Several chemically similar Δ9-THC derivatives (e.g., classical cannabinoids) have also been developed. Cesamet® (nabilone) and HU-210 are examples of these analogs and exemplify how slight modifications of the Δ9-THC chemical structure can significantly alter potency (Figure 3A). The anti-emetic nabilone approaches Δ9-THC in potency and is the only Δ9-THC analog that has ever been approved in the US. HU-210 has never been approved for medical use primarily because it is 100- to 800-fold more potent than Δ9-THC.
Nonclassical Cannabinoids
Nonclassical cannabinoids, or cannabinoids that are chemically distinct from Δ9-THC (Figure 3B), have also been explored for potential therapeutic use. From the 1960s through the 1980s, scientists at Pfizer developed Δ9-THC analogs known as cyclohexylphenols (91, 98) and designated as “CP” compounds. In general, these drugs have a simplified chemical structure consisting of two of the three ring structures of Δ9-THC. Several derivatives as well as their alkyl homologs have been developed. These include the compounds CP-50,556-1, CP-47,497, and the C8 akyl homolog of CP-47,497 [(C8)-CP-47,497]. CP-50,556-1, also known as levonantradol, is one of the original analogs shown to be a very effective analgesic and anti-emetic for use in patients undergoing chemotherapy (91). The water solubility of levonantradol makes it amenable for drug formulations and delivery, but adverse side effects prevented eventual FDA approval. CP-47,497 and (C8)-CP-47,497 are at least thirtyfold more potent than Δ9-THC (99).
Aminoalkylindoles (AAIs)
Pravadoline is another nonclassical cannabinoid that bears little resemblance to Δ9-THC but is typically further classified as an aminoalkylindole (Figure 3C) (22). Research scientists at Sterling Drug Company developed pravadoline as an analgesic not associated with gastric irritation (91, 98). The drug has clinical effects similar to those of Δ9-THC and a higher relative potency. Receptor binding assays determined that pravadoline, and similar compounds like WIN-55212-2, bind CBRs and act as cannabinoids (91, 100). Further studies demonstrated that these compounds retain high affinity for both CB1Rs and CB2Rs, produce behavioral effects similar to those produced with Δ9-THC, have cannabimimetic discriminative stimulus effects in rats and rhesus monkeys, and their effects are attenuated with cannabinoid antagonists (e.g., SR-141716A) (101).
J. W. Huffman, a chemist at Clemson University, also synthesized and characterized other AAIs. These compounds, designated by the initials “JWH”, include several naphthoylindoles, naph-thoylpyrroles, and other related structures (98, 101, 102). JWH compounds show differential selectivity toward CB1Rs and CB2Rs. For example, JWH-018 and JWH-073 bind both receptors with differing affinities, whereas JWH-015 appears to bind only CB2Rs with high affinity. The pharmacological profiles of the JWH compounds are similar to those of Δ9-THC, with JWH-018 having greater relative potency (101, 103–113).
Use as Designer Drugs in Europe and United States: Compounds in K2/Spice
Major limitations in the development of new cannabinoids have been undesirable psychoactive properties and public perception of cannabinoid use. Recreational drug users, however, have renewed the interest in several historical cannabinoids. For obvious reasons, these individuals have identified cannabinoids that bind and mediate responses through CB1Rs, that are not regulated through existing statutes, and that are not detectable by common drug screens (114). Several of these compounds became available through local head shop and internet resources in the early to mid-2000s (Figure 1), and in 2004, a wide variety of products marketed as “legal” smoking blends began gaining popularity worldwide. “Spice” and “K2” represent two of the more popular brand names that became widely available in the United States (Figure 4). They are commonly ingested by smoking, although these products are typically labeled as “incense” and “not for human consumption” or “for aromatherapy only.” Recent forensic determinations in Europe and the United States found that many of these herbal products are laced with HU-210, JWH-018, JWH-073, CP-47,497, and (C8)-CP-47,497 (102, 115–120). Derivatives, such as JWH-015, that show selectivity for CB2Rs have not been detected in these herbal products, which supports the notion that these products are indeed intended for their psychoactive properties despite product labeling.

An example of K2 packaging.
Recognition of K2/Spice Intoxication and Clinical Implications
Of concern to clinicians and public health officials is the lack of clinical information related to the pharmacology of these compounds in man. It is expected that increased potencies exhibited by many of these synthetic agonists will lead to longer durations of action and an increased likelihood of adverse effects. In addition to the active ingredients, the plant material used for manufacturing (Table 2) (as well as varying product mixtures) may lead to other unexpected toxic outcomes (102). Existing clinical literature suggests that severe and life-threatening symptoms (Table 3) may occur in users that naïvely consider these products to be “marijuana substitutes” (121–124).
Plants Commonly Used to Produce Herbal Products Laced with Cannabinoids
Adverse Clinical Effects Reported with Use of K2 Products
Unlike the low efficacy partial agonist effects of Δ9-THC, several of the AAIs used to produce these products are characterized as full agonists with increased potency. To date, two deaths have been reported in the United States with K2 product use. One involved an adolescent who committed suicide after experiencing extreme anxiety following use of these compounds (125). The second involved an adolescent who died following a coronary ischemic event (126). Although suicidal tendencies have been associated with the use of CB1 antagonists (e.g., rimonabant), additional data are needed to elucidate the mechanisms for clinical toxicity in humans. The lack of a definitive diagnostic test for K2 compounds also hinders the recognition and appropriate medical treatment of side effects from these products. It is anticipated that reportable injuries and deaths will significantly increase as detection, and thus recognition, improve.
Need for Regulation of K2/Spice Compounds
Very little regulation controlling the manufacture, distribution, and use of K2 products existed when these products emerged in the United States. One of the more potent derivatives, HU-210, was listed as a Schedule I substance when authorities discovered its use. CP and JWH compounds did not fall under existing regulations because of their structural dissimilarity with Δ9-THC (Figure 3). Several European countries first began regulating these products in 2009 (102), while United States regulations started in early 2010 (Figure 1). Kansas, Kentucky, Missouri, Georgia, Alabama, and Arkansas were among some of the first states to take legislative action through state and local statutes and rules. Several other states, local municipalities, and the United States Armed Forces have also adopted similar regulations banning these substances. Pursuant to the temporary scheduling provisions under 21 U.S.C. 811(h) of the Controlled Substances Act, the United States Drug Enforcement Agency has initiated the process for scheduling JWH-018, JWH-073, JWH-200, CP-47,497, and (C8)-CP47,497 as Schedule I substances.
Detection and Monitoring of K2/Spice Compounds
The renewed interest in synthetic cannabinoids and their increased use as designer drugs raises significant public health concerns. Basic and clinical research is needed to address these concerns. A definitive diagnostic is needed to help associate clinical symptoms with specific drug use and to help serve as a deterrent for users seeking to avoid detection. Additional testing for other drugs of abuse will help to delineate K2 specific symptoms from those associated with the use of other illicit drugs. Understanding the clinical pharmacology of these substances will allow scientists to discern the relative toxicity associated with varying synthetic can-nabinoid mixtures and routes of administration. This information is vital for the future education of communities, physicians, and lawmakers as they work together to protect public health.
Conclusions
Beneficial Therapeutics or Designer Drugs of Abuse?
Although marijuana has been used (and abused) for centuries, the discovery that Δ9-THC activates CB1Rs resulted in a relatively recent push for development of CB1R agonists and antagonists for clinical use. Unfortunately, therapeutic doses of both CB1R agonists and antagonists produce unacceptable psychiatric effects, severely limiting future drug discovery. Several strategies to mitigate these side effects are being investigated, including development of CB1R ligands with restricted CNS access, identifying allosteric modulators of CB1Rs, indirect enhancement of CB1R activity by blocking endocannabinoid breakdown, and use of neutral CB1 antagonists.
Concurrent with attempts to produce highly selective and potent CB1R ligands for improved therapeutic use, a novel class of drugs of abuse emerged known as K2 or Spice. These drugs produce psychotropic actions via CB1R activation; however, because of their structural dissimilarity to Δ9-THC and inconsistent regulation, they are touted as “legal” forms of marijuana. Importantly, owing to unique adverse effects relative to Δ9-THC, users experimenting with K2 products are exposed to drugs that are extremely variable in both composition and potency, and they often develop serious adverse effects. Thus, an understanding of the clinical pharmacology of these substances is needed to allow scientists to discern their relative toxicity. Further, this information is vital for the future education of communities, physicians, and lawmakers as they work together to protect public health.
Several hurdles remain for developing selective CB1R ligands with tolerable adverse effects in patients. Complicating matters further is the recent discovery that several experimental compounds are being used as designer drugs capable of avoiding detection and regulation. Although there are significant public health problems associated with any drug of abuse, the information reviewed here indicates that the potential therapeutic benefits of cannabinoids outweigh the negative aspects. Therefore, we conclude that marijuana-based drugs are indeed worthy of future study and characterization, in order to rigorously investigate their pharmacodynamic and potential clinical effects.
Acknowledgments
The Authors would like to thank Ms. Vi-Huyen Le and Ms. Kristen Richmond for their technical assistance while preparing this manuscript. The authors would also like to thank Ms. Cindy L. Moran with the Arkansas State Crime Laboratory for providing photographs of K2 Products.
This work was supported, in part, by the Association of Public Health Laboratories [Grant Innovations in Quality Public Health Laboratory Practice] (JHM) and the Centers for Disease Control [Grant U90/CCU616974-07 and Contract 200-2007-21729] (JHM). In addition, this work was supported by a pilot grant awarded to LPJ through the Arkansas Center for Clinical and Translational Research, which is funded by the National Center for Research Resources [1 UL 1RR029884, Curtis Lowery, PI].
The content is solely the responsibility of the authors and does not necessarily represent the official views of the Association of Public Health Laboratories, Centers for Disease Control, National Center for Research Resources, or the National Institutes of Health.
Footnotes
-
Authorship contributions:
Wrote or contributed to the writing of the manuscript: Seely K.A., Prather, P.L., James L.P., and Moran, J.M. all contributed equally in writing and preparing this manuscript.
-
Other: Moran, J.M. and James, L.P. acquired funding for the research.
- Copyright © 2011
References
Kathryn A. Seely, PhD, (left) received her BS in biology from Christian Brothers University in Memphis, Tennessee, before completing her doctorate at the University of Arkansas for Medical Sciences in Dr. Prather’s laboratory. Currently, she is a post-doctoral fellow at the University of Arkansas for Medical Sciences, investigating novel treatments for sepsis-induced acute kidney injury.
Paul L. Prather, PhD, (second from left) is an Associate Professor in the Department of Pharmacology and Toxicology at the University of Arkansas for Medical Sciences, College of Medicine. His NIH-funded research interests involve understanding the cellular and molecular mechanisms of signal transduction mediated by G protein–coupled receptors with which drugs of abuse interact. In particular, the research focus of his laboratory involves study of drugs of abuse that signal through opioid (μ-, δ-, and κ-) and cannabinoid (CB1 and CB2) receptors.
Laura James, MD, (second from right) is Section Chief of Clinical Pharmacology and Toxicology at Arkansas Children’s Hospital and is a Professor of Pediatrics at the University of Arkansas for Medical Sciences. She has NIH-funded research programs focusing on acet-aminophen toxicity in humans and in experimental models. Her expertise extends to the performance of pediatric pharmacokinetic and pharmacodynamic studies in children.
Jeffery H. Moran, PhD, (right) is Branch Chief of Environmental Chemistry at the Arkansas Department of Health, Public Health Laboratory. He also holds a faculty appointment in the College of Medicine at the University of Arkansas for Medical Sciences, Department of Pharmacology and Toxicology. His research funding by the Center for Disease Control (CDC) focuses on xenobiotic metabolism and analytical detection. He also provides added expertise for public health programs. E-mail jeffery.moran{at}arkansas.gov; fax (501)-661-2972.


