New Therapeutic Approaches to Combat Arterial Thrombosis
Better Drugs for Old Targets, Novel Targets, and Future Prospects
Abstract
Cardiovascular disease and stroke are predominant causes of death in developed countries. Rupture of atherosclerotic plaque in an artery wall and the ensuing thrombotic events are the triggers for acute ischemic injury in these diseases. Platelet activation and aggregation play key roles in this process of atherothrombosis. Anti-platelet drugs thus provide the primary therapeutic strategy to combat these diseases. Dual therapy with aspirin and clopidogrel is the current standard of care for most patients, but it has significant limitations. This provides an impetus for developing new anti-platelet drugs. One new drug has received FDA approval recently; prasugrel targets the platelet P2Y12 receptor, just like clopidogrel. Several other new drugs are showing great promise in clinical trials and appear to be nearing approval. Some of these drugs have traditional targets on the platelets; others, such as vorapaxar, terutroban, and sarpogrelate, generate more excitement as they are directed against novel targets.
Introduction
Coronary artery disease is a major health problem in developed countries. Deposition of atherosclerotic plaque in the arterial wall is the primary event in pathogenesis of this disease. It results in arterial stenosis and impaired relaxation of the smooth muscle lining of the vessel. Analogous events occur in cerebrovascular disease and peripheral artery disease. Severe arterial stenosis may, in itself, compromise the function of the heart, brain, or another organ through ischemia. It is the rupture of a vulnerable portion of atherosclerotic plaque and the ensuing thrombotic events, however, that are largely responsible for both the irreversible injury to tissues and the mortality from these diseases.
Platelet activation and aggregation play a predominant role in atherothrombosis—that is, the formation of a thrombus as a result of the rupture of an atherosclerotic plaque (1–3) (Figure 1). Platelets also participate in the initiation and progression of an atherosclerotic lesion (4, 5). Plaque rupture causes exposure of the subendothelial proteins, including von Willebrand factor and collagen. Platelets adhere to these exposed proteins and become activated through signaling mechanisms that are engaged directly (3). This process is amplified extensively as the activated platelets generate and release soluble platelet agonists, ultimately leading to aggregation of the platelets and formation of a thrombus that may occlude the artery (6–8). Acute coronary syndrome is a catch-all term that encompasses the clinical symptoms of such an occlusion if it occurs in the heart (9). Complete occlusion results in myocardial infarction. A near-complete or brief cerebrovascular occlusion may trigger a transient ischemic attack, whereas prolonged occlusion results in ischemic stroke (10).
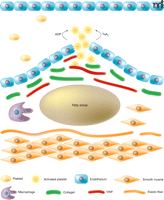
Invasion by macrophages and migration of smooth muscle cells leads to deposition of foam cells to form a fatty streak beneath the subendothelium. Rupture of this atherosclerotic plaque may ultimately occur and expose the proteins of the subendothelial matrix. Platelets adhere to von Willebrand factor (VWF) and collagen in the exposed subendothelium. The resulting platelet activation is amplified through paracrine signaling by ADP and thromboxane A2. Such signals are the target of most of the current anti-platelet drugs.
The paracrine signaling mechanisms that amplify platelet activation are the primary targets for the drugs currently used in acute treatment of atherothrombosis (11). The same drugs are also used for secondary prophylaxis to prevent further episodes of thrombosis. Two key events in platelet activation are the generation of thromboxane A2 and the secretion of the contents from platelet granules (6). Signaling mechanisms in the platelet characteristically trigger a rise in intracellular calcium concentration and cause activation of phospholipase A2. Arachidonic acid released by cleavage of phospholipids in the platelet membrane serves as a substrate for cyclo-oxygenase, leading to the generation of thromboxane A2. As this eicosanoid is itself a strong platelet agonist, this process is a major contributor to the paracrine signaling mechanisms responsible for amplifying platelet activation (6). Aspirin (acetylsalicylic acid) irreversibly inhibits cyclo-oxygenase and thereby blocks this paracrine mechanism. ADP is one of the main constituents of δ-granules released upon platelet activation. It also plays a key role in the paracrine signaling mechanisms that amplify the platelet response (6, 7). Clopidogrel is a prodrug with an active metabolite that irreversibly antagonizes the P2Y12 receptor, one of two classes of ADP receptor on the platelet. The P2Y12 receptor plays a predominant role in the positive feedback mechanisms for platelet activation (7). Clopidogrel blocks this pathway for paracrine signaling that leads to platelet activation.
Limitations of Current Therapeutic Agents
Dual anti-platelet therapy with aspirin and clopidogrel is currently the standard of care for prevention of adverse cardiovascular events in most patients at high risk owing to a prior myocardial infarction or recent placement of a stent (11, 12). One or both drugs are also used therapeutically in most patients with acute coronary syndrome and are used prophylactically after a transient ischemic attack or ischemic stroke. Nonetheless, there are significant limitations to the use of these drugs in coronary artery disease and cerebrovascular disease. First, the two drugs, used in what is thought to be an optimal combination, reduce the risk of a serious vascular event (myocardial infarction or ischemic stroke) only to a modest extent (typically about 25%) in patients with cardiovascular disease. One factor that may contribute to the inadequate therapeutic efficacy is the purported phenomenon of resistance to each drug (i.e., pharmacological ineffectiveness). The existence of other mechanisms for platelet activation and aggregation is undoubtedly also a contributory factor. Second, both drugs are associated with an increased risk of bleeding, particularly when used in combination. One of the most difficult challenges in antithrombotic therapy is achieving an optimal balance between a reduced risk of thrombosis and an increased risk of bleeding (11, 13, 14). An increase in the risk of bleeding is virtually inevitable with any anti-platelet drug (as with any anticoagulant), but the risk–benefit balance should always be favorable for any therapeutic strategy.
Aspirin
A low dose of aspirin (75–100 mg per day) is characteristically chosen for anti-platelet therapy (15). This strategy takes advantage of the anucleate nature of the platelet and the irreversible action of aspirin in acetylating a serine residue in the active site of cyclo-oxygenase 1. Low-dose aspirin typically causes complete inhibition of cyclo-oxygenase in the platelet with minimal inhibitory effect on cyclo-oxygenase in the vascular endothelial cells. The latter cells are a source of prostacyclin, which suppresses platelet function and is particularly important in high-shear regions of the vasculature, such as the coronary arteries (16). A low dose of aspirin is also advantageous in minimizing the incidence of adverse effects on the gastrointestinal tract (15).
Aspirin has proven effective at reducing the incidence of myocardial infarction and stroke in patients at high or intermediate risk (17). Because it appears to be less effective in some patients, however, the concept of “aspirin resistance” has emerged (18, 19). Those patients with less than complete suppression of thromboxane A2 generation do appear to be at significantly higher risk for an adverse vascular event (1, 19). What is less clear, however, is whether these patients are truly resistant to the action of aspirin on the cyclo-oxygenase enzyme or whether there is another explanation (18). Non-compliance with the drug regimen appears to contribute significantly to the prevalence of apparent resistance to aspirin, but other pathways for generation of thromboxane A2 may also contribute.
Clopidogrel
Like aspirin, clopidogrel has proven effective at reducing the incidence of myocardial infarction and stroke in patients at risk (20). Clopidogrel causes irreversible blockade of the P2Y12 receptor on the platelet, thereby substantially suppressing the amplification of platelet activation that occurs through the synergistic action of ADP released from the platelet δ-granules (7, 21). As aspirin and clopidogrel have distinct targets, the two drugs are often used in combination to cause more complete suppression of platelet function (22). This results in greater efficacy from a clinical perspective, albeit with a greater incidence of bleeding complications and adverse effects on the gastrointestinal tract.
Substantial variability in the response to clopidogrel is a major limitation of this drug (18, 23, 24). As with aspirin, a lack of compliance with the drug regimen can contribute. Pharmacokinetic factors, however, probably have a significant role in causing true resistance to the action of clopidogrel. A significant problem is that conversion of clopidogrel to its active metabolite is inefficient (11, 18). The prodrug is transformed through two reactions that are catalyzed by isozymes of cytochrome P450 (Figure 2A). Each step in this transformation occurs in competition with an esterase-mediated hydrolysis that yields an inactive metabolite, and these esterase-mediated pathways tend to predominate. Indeed, only about 15% of the pro-drug is typically metabolized to the active metabolite, and 85% is converted to inactive metabolites. Polymorphic variation in some of the cytochrome P450 isozymes contributes to the variability in efficacy of clopidogrel between patients (25, 26). This applies particularly to CYP2C19, which normally plays a predominant role in the first step in transformation of clopidogrel (yielding 2-oxo-clopidogrel) and also contributes to the second step in transformation to the active metabolite. There are two non-functional variants of CYP2C19 (designated CYP2C19*2 and CYP2C19*3) that occur with significant prevalence in some ethnic groups. A reduced efficacy of clopidogrel has been documented in patients heterozygous or homozygous for the non-functional CYP2C19*2 variant, both in terms of platelet responsiveness to ADP ex vivo and in terms of clinical outcome (i.e., adverse cardiovascular events) (27, 28). Other drugs are purported to reduce the efficacy of clopidogrel through inhibition of cytochrome P450 (18). A potential interaction with omeprazole is particularly pertinent, as it is often used to limit the adverse gastrointestinal effects of clopidogrel, but studies on this topic have yielded conflicting data (11, 26). There is also evidence to suggest that polymorphic variation in the ATP-binding cassette B1 gene might influence the efficacy of clopidogrel through a change in its absorption from the intestine (29).
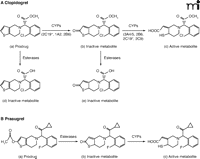
A. Clopidogrel metabolism from the prodrug form to its active metabolite occurs through two reactions that are mediated by cytochrome P450. Competing pathways for inactivation by esterases are also shown. B. Prasugrel metabolism occurs through two steps, one mediated by esterases and the other by cytochrome P450.
Ticlopidine is an alternative to clopidogrel that is rarely used because it has a greater tendency to cause severe blood dyscrasias, including neutropenia and aplastic anemia (30). Its structure and mechanism of action are similar to clopidogrel. It is also a prodrug that requires transformation to an active metabolite by cytochrome P450. As ticlopidine lacks the methyl ester moiety present in clopidogrel, however, it is not inactivated by esterases and may be transformed more consistently to its active metabolite.
Glycoprotein IIb–IIIa Antagonists
Regardless of how platelet activation is initiated, aggregation of the platelets arises through a common pathway that involves binding of fibrinogen (and other ligands) to glycoprotein IIb–IIIa (GpIIb–IIIa, integrin αIIbβ3) on the activated platelets (21). Antagonists of GpIIb–IIIa thus provide an additional strategy to suppress formation of a thrombus. Three such antagonists (abciximab, eptifibatide, and tirofiban) are beneficial as short-term adjuncts to the other anti-platelet agents during percutaneous coronary intervention, particularly in high-risk patients (31, 32). These particular agents require intravenous administration. Although orally active GpIIb–IIIa antagonists have been developed, they have not shown a satisfactory benefit–risk profile (32, 33).
Dipyridamole
Prophylaxis to prevent a second occurrence of ischemic stroke differs somewhat from that to prevent a second myocardial infarction (34). Dual anti-platelet therapy with aspirin and clopidogrel engenders too great a risk of hemorrhage in stroke patients owing to differences in the endothelial cells in the cerebral vasculature. Combination therapy with aspirin and dipyridamole is used as an alternative to single-drug therapy with aspirin or clopidogrel in such patients (33). Dipyridamole is an inhibitor of phosphodiesterase and suppresses platelet function primarily by elevating the concentration of cyclic GMP (35).
Better Drugs for Old Targets on the Platelet
There have been substantial efforts in recent years to develop new anti-platelet drugs. Most progress has been made in developing drugs that target the same entities on the platelet as current drugs but overcome some of the limitations of those drugs. One area of particular emphasis has been new drugs that target the P2Y12 receptor. Refinements are also occurring for drugs with other targets.
Prasugrel
Given that a major limitation of clopidogrel is its inefficient transformation to the active metabolite, one strategy to develop a better P2Y12 antagonist is to design a prodrug with a more direct and efficient means of transformation to its active metabolite (36). This strategy led to the development of prasugrel (Figure 2B). Key structural features that define interaction with the P2Y12 receptor are analogous to those of clopidogrel. The structure has been modified, however, to facilitate formation of the thiolactone intermediate (akin to 2-oxo-clopidogrel) through rapid hydrolysis by an esterase (37). Moreover, the methyl ester group that is sensitive to esterase cleavage in clopidogrel has been replaced to preclude such cleavage. As a result of these structural changes, transformation of prasugrel to its active metabolite requires catalysis by cytochrome P450 at only a single step (36, 37). As multiple isozymes of cytochrome P450 can effect this transformation, it occurs consistently and efficiently in individuals with different polymorphic variants of these isozymes (29, 37). The active metabolite of prasugrel contains a sulfhydryl moiety that reacts covalently with a cysteine residue on the P2Y12 receptor. As the inhibition is irreversible, receptor function is suppressed for the life span of the platelet.
Clinical trials have demonstrated that prasugrel is substantially more potent than clopidogrel in terms of blockade of ADP-induced platelet function ex vivo (38, 39). Moreover, those individuals that respond poorly to clopidogrel show robust inhibition of platelet function with prasugrel (39). The greater potency for prasugrel can be attributed simply to its more efficient transformation to the active metabolite. More importantly, large-scale randomized trials have documented a more favorable clinical outcome with prasugrel than with clopidogrel in patients with acute coronary syndrome that is remedied by percutaneous coronary intervention (40, 41). Prasugrel received FDA approval in July 2009 for this clinical use. The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel – Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38) showed a reduction of 19% in the relative risk of serious adverse vascular events in the patients receiving prasugrel (42). The finding of a 52% reduction in risk of stent thrombosis with prasugrel was particularly notable (43). More detailed analysis of the data from this trial has indicated that the use of prasugrel may be particularly beneficial in some groups of patients, such as those with diabetes mellitus (44). In contrast, there was no benefit in patients with a history of stroke or transient ischemic attack; such patients also had an increased risk of hemorrhage (42). Prasugrel also failed to achieve a favorable benefit–risk profile at the dosage used in patients aged ≥ 75 years and in those of body weight < 60 kg.
The clinical benefit of prasugrel over clopidogrel was counterbalanced to some extent in the TRITON-TIMI 38 trial by an increased risk of bleeding (42). There is also a concern that prasugrel might increase the incidence of some cancers (45). The greater risk of bleeding is an inevitable consequence of the more effective blockade of the platelet P2Y12 receptor in patients treated with prasugrel (46). It may also contribute to the lack of clinical benefit in patients with a history of stroke or transient ischemic attack. A reduction in the dose of prasugrel can presumably offset the increased risk of bleeding and might then make prasugrel the preferred drug even in the latter patients. Prasugrel should maintain the advantage over clopidogrel of more consistent transformation to its active metabolite regardless of the particular group of patients under treatment.
Ticagrelor, Cangrelor, and Elinogrel
The rationale in developing prasugrel was to design a prodrug that is converted by a simpler and more efficient pathway to its active metabolite. A divergent strategy has been employed to design three further antagonists of the platelet P2Y12 receptor: ticagrelor, cangrelor, and elinogrel (11, 46). These agents are direct acting antagonists that do not require metabolic transformation from a prodrug. Moreover, all three are reversible antagonists of the P2Y12 receptor.
Ticagrelor (AZD6140) is an orally active, direct acting, reversible antagonist of the platelet P2Y12 receptor (47, 48). It appears to have the most promise among this new class of antagonists. Ticagrelor is structurally distinct from the thienopyridine drugs (i.e., clopidogrel, ticlopidine, and prasugrel); it has greater structural similarity to adenosine (Figure 3A). Ticagrelor has a rapid onset of action after oral administration, displaying maximal suppression of platelet function within about 2 hours. The degree of blockade of the platelet P2Y12 receptors can be titrated by adjusting the dosage of ticagrelor; near-complete blockade can be achieved if warranted (49). As the drug is a reversible antagonist with a relatively short plasma half-life of 12 hours, platelet function recovers quite rapidly on discontinuation of the drug. This provides at least a theoretical advantage for ticagrelor over both clopidogrel and prasugrel in the event that a patient requires urgent surgical intervention, such as a coronary artery bypass graft.
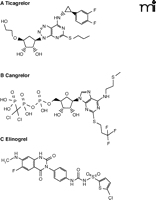
The varied structures of (A) ticagrelor, (B) cangrelor, and (C) elinogrel are shown. All three drugs share the same molecular target, the platelet P2Y12 receptor.
Clinical trials have provided evidence that ticagrelor may have a better benefit–risk profile than clopidogrel in patients with acute coronary syndrome. The Platelet Inhibition and Patient Outcomes (PLATO) trial showed a reduction of 16% in the relative risk of death from vascular causes in the patients receiving ticagrelor (50, 51). As had been found with prasugrel, ticagrelor also proved to be markedly superior to clopidogrel in preventing thrombosis within a stent. There was little, if any, increase in the risk of bleeding to offset the benefit of ticagrelor in terms of clinical outcome. In the patients receiving ticagrelor, however, there was an increased incidence of two other adverse effects, dyspnea and ventricular pauses, particularly during the first week of treatment (51). Ticagrelor received approval by the European Commission in December 2010 for use in the European Union, but it has not currently received FDA approval. The finding of an inexplicable difference in the clinical outcome during the PLATO trial between patients in North America and those in Europe may have been a contributory factor in the divergence in approval (52) (Box 1).
Why Did the PLATO Trial Show Regional Differences in Outcome?
The PLATO trial involved studies on 18,624 patients in 43 countries. Ticagrelor demonstrated a 16% reduction in the primary end point (cardiovascular death, myocardial infarction, or stroke) compared with clopidogrel in the entire study. Patients in the United States, however, seemed to have a worse outcome on ticagrelor than on clopidogrel, although the hazard ratio was not significantly greater than 1.0. The difference among regions was significant (p < 0.05).
The study included relatively few (1,413) patients from the United States. These patients differed in several notable ways from those in other regions. The most marked difference was in the median maintenance dose of aspirin (325 mg in the United States and 100 mg in other regions). Statistical analysis revealed a strong interaction between the aspirin maintenance dose and the effectiveness of ticagrelor throughout all regions (p < 0.0001). Patients on low-dose aspirin in the United States fared just as well on ticagrelor as those in other regions.
The Cardiovascular and Renal Drugs Advisory Committee of the FDA were not persuaded by the aforementioned evidence of influence by the aspirin maintenance dose, yet they recommended approval of ticagrelor by a 7:1 vote. The FDA was not obliged to follow the recommendation of the Committee and chose not to approve the drug.
Cangrelor (AR-C69931MX) has some structural and functional similarities to ticagrelor (Figure 3B). It is a structural analog of ATP that requires intravenous administration, unlike ticagrelor (53). Cangrelor acts virtually instantaneously when administered as a bolus. It also has a very short plasma half-life of 7 minutes owing to its rapid dephosphorylation; as a result, platelet function recovers fully in less than 1 hour on discontinuation of the drug. Clinical trials have established the safety of cangrelor. The two Cangrelor versus Standard Therapy to Achieve Optimal Management of Platelet Inhibition (CHAMPION) trials that involved a head-to-head comparison with clopidogrel, however, have been discontinued owing to a perceived lack of clinical benefit with cangrelor (46, 54, 55). It appears, nonetheless, that cangrelor might have a place in the therapeutic armamentarium to combat arterial thrombosis if it is used on a short-term basis in patients that are awaiting surgical intervention and have had treatment with clopidogrel (or prasugrel) stopped to preclude excessive bleeding. The Maintenance of Platelet Inhibition with Cangrelor after Discontinuation of Thienopyridines in Patients Undergoing Surgery (BRIDGE) trial to assess the value of cangrelor as a rapidly reversible bridging agent in this setting is currently ongoing (11).
Elinogrel (PRT060128) is the most recent addition to the new class of direct acting and reversible P2Y12 receptor antagonists (Figure 3C), at least in terms of agents that have entered clinical trials. Its unique feature is that it is suitable for either oral or intravenous administration (56). This property should facilitate transition from acute use where a high degree of platelet inhibition is warranted, such as during percutaneous coronary intervention, to chronic use with less suppression of platelet function after intervention. Elinogrel has essentially an immediate onset of action with intravenous administration and shows a fairly rapid recovery of platelet function after discontinuation of the drug. A phase 2 clinical trial (57) has demonstrated the potential value of elinogrel as an anti-platelet agent, and plans are under way for a phase 3 trial to start later in 2011.
Other reversible antagonists of the platelet P2Y12 receptor, such as BX 667, are currently at the pre-clinical stage of drug development (58, 59).
Cilostazol
Elevation of cyclic AMP or cyclic GMP within the platelet is an effective means to suppress platelet function. Cilostazol is a selective inhibitor of the phosphodiesterase type 3 family (60). It suppresses platelet function and also promotes vasodilation. Cilostazol has FDA approval for treating the symptoms of intermittent claudication (i.e., pain and cramping in the lower leg) as a result of peripheral artery disease. Several small-scale clinical trials have been conducted to assess the value of cilostazol in the treatment of ischemic stroke. The trial results suggest that cilostazol may have a better benefit–risk profile than aspirin in stroke patients as it has a lesser tendency to cause a hemorrhagic stroke. Cilostazol may also be useful as a third anti-platelet drug to prevent restenosis and thrombosis in stents (61). Nonetheless, larger-scale trials are needed to confirm this finding.
Nitroaspirin
Nitric oxide suppresses platelet function through an elevation of cyclic GMP. This provided the primary rationale to develop nitroaspirin (NCX 4016), a drug that comprises a nitric oxide-releasing moiety covalently linked to aspirin (62, 63). The dual actions of this drug provide synergistic suppression of platelet function (64). This property is complemented by a direct inhibitory effect of the released nitric oxide on atherosclerosis and its ability to protect the gastric mucosa from injury. Small-scale clinical trials have suggested that nitroaspirin has beneficial effects in patients with intermittent claudication (65). Larger-scale trials are needed to determine whether it will have a place in the clinical repertoire of drugs used to combat atherothrombosis.
Novel Targets for Anti-Platelet Drugs
Although much of the effort to develop new anti-platelet drugs has focused on targets that are already well established, there has also been considerable progress in developing drugs that inhibit novel targets on the platelet (Figure 4). In terms of progress through clinical trials, a major focus has been on drugs that target the platelet receptors for thromboxane A2, thrombin, and serotonin.
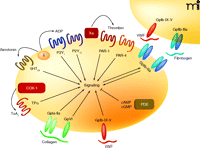
The current anti-platelet drugs target cyclo-oxygenase 1 (COX-1), the P2Y12 receptor, platelet glycoprotein IIb–IIIa (GpIIb–IIIa), and phosphodiesterase (PDE). Many of the drugs under development have new targets on the platelet, including the thromboxane A2 receptor subtype α (TPα), PAR-1 receptor for thrombin, 5-HT2A receptor for serotonin, and platelet glycoprotein Ib–IX–V (VWF receptor) and glycoprotein VI (collagen receptor).
Thromboxane A2 Receptor
Blockade of the thromboxane A2 receptor has two potential advantages over the suppression of thromboxane A2 generation as an antithrombotic strategy. First, blockade of the receptor precludes its activation not only by thromboxane A2 but also by other eicosanoids, such as isoprostanes, that are generated non-enzymatically (66). In contrast, aspirin suppresses only the enzymatic pathway that leads to generation of thromboxane A2. Secondly, thromboxane A2 is not generated exclusively in platelets, nor are its receptors restricted to platelets. Receptors for thromboxane A2 are present on vascular endothelial cells and smooth muscle cells. These receptors contribute to vasoconstriction and to the progression of atherosclerotic lesions. Despite these theoretical advantages, an earlier acute study in patients with myocardial infarction showed no difference in clinical out-come between aspirin and ridogrel, a combined thromboxane A2 synthase inhibitor and receptor antagonist (67).
Terutroban (S18886) is a reversible antagonist of the thromboxane A2 receptor (Figure 5A). Pre-clinical studies have demonstrated the effectiveness of terutroban at suppressing arterial thrombosis and have suggested that it is less prone to cause bleeding (68, 69). A small-scale clinical trial has indicated that terutroban may be more effective than aspirin at restoring endothelial function in patients with coronary artery disease (70). Another small-scale study has suggested that the drug may be effective at secondary prevention of ischemic stroke (71). The latter outcome prompted the Prevention of Cerebrovascular and Cardiovascular Events of Ischemic Origin with Terutroban in Patients with a History of Ischemic Stroke or Transient Ischemic Attack (PERFORM) trial, a large-scale phase 3 trial comparing terutroban against aspirin for secondary prevention of stroke (72). This trial was recently discontinued near to its projected end because terutroban appeared to offer no benefit over aspirin.
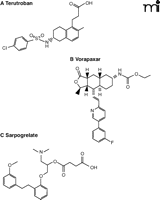
Structures are shown for (A) terutroban, an antagonist of the thromboxane A2 receptor; (B) vorapaxar, a PAR-1 antagonist; and (C) sarpogrelate, an antagonist of the serotonin 5-HT2A receptor.
Another strategy for blockade of the thromboxane A2 receptor that is currently the subject of pre-clinical studies is use of an anti-diabetic drug, glibenclamide (73).
Thrombin Receptor PAR-1
Aside from its prominent role in the coagulation cascade, thrombin is a very potent platelet agonist (74, 75). Moreover, platelet activation by any agonist leads to phosphatidylserine exposure on the platelet membrane, creating a procoagulant surface that triggers the generation of thrombin. There are two distinct receptors for thrombin on the platelet, both members of the protease-activated receptor (PAR) family. Each of these two receptors, PAR-1 and PAR-4, is activated through enzymatic cleavage by thrombin. The two receptors then act in concert to induce a plethora of signals that result in platelet activation. PAR-1 appears to play the predominant role, so it has been selected as the preferred target for new anti-platelet drugs.
Vorapaxar (SCH 530348) is a selective and competitive antagonist of the PAR-1 thrombin receptor that is based on the natural product himbacine (76, 77) (Figure 5B). It has no effect on the proteolytic cleavage by thrombin of fibrinogen into fibrin. Phase 2 clinical trials have shown that vorapaxar provides effective blockade of PAR-1 function ex vivo and have established that it does not cause significant bleeding (77). Two large-scale phase 3 trials, entitled Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRA-CER) and Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2°P–TIMI 50), were initiated to assess whether it is beneficial to add vorapaxar to the standard therapeutic care in patients with acute coronary syndrome or as a means for secondary prevention of atherothrombotic ischemic events (8, 74). In both trials, treatment with vorapaxar has been discontinued in patients with a history of stroke because the drug appears to increase the risk of intracranial bleeding. The TRA 2°P trial continues, nonetheless, with its focus adjusted toward patients with coronary or peripheral artery disease.
A second PAR-1 antagonist, atopaxar (E5555), has also been developed. Small-scale clinical trials suggest that it also has promise as a potential anti-platelet agent (78, 79). Larger-scale phase 3 trials, however, are currently awaited. Other PAR-1 antagonists are also in the drug development pipeline.
Serotonin 5-HT2A Receptor
Platelet activation triggers release of a number of substances from the δ-granules. The synergistic effect of released ADP has been a major focus of research into platelet function and development of anti-platelet drugs, whereas other released substances have often been disregarded. One such substance is serotonin (5-hydroxytryptamine, 5-HT). Just like ADP, serotonin contributes to the positive feedback mechanism that amplifies platelet activation once it has been initiated. Serotonin triggers activating signals in the platelet through the 5-HT2A subtype of serotonin receptor (80). It also acts through the same receptor to promote contraction of vascular smooth muscle.
Sarpogrelate (MCI-9042) is a selective and reversible antagonist of the 5-HT2A receptor (Figure 5C). Phase 2 studies have demonstrated the safety of sarpogrelate for treatment of peripheral artery disease and suggested that it may reduce the symptoms of intermittent claudication (81). It is used extensively to treat peripheral artery disease in Japan, China, and the Republic of Korea. The Sarpogrelate-Aspirin Comparative Clinical Study for Efficacy and Safety in Secondary Prevention of Cerebral Infarction (S-ACCESS) trial compared the efficacy of sarpogrelate and aspirin for secondary prevention of stroke (82). Serious adverse vascular events occurred with similar frequency in patients receiving both drugs, whereas bleeding complications were less prevalent in those treated with sarpogrelate. Subgroup analysis of the trial data suggested that sarpogrelate may be more effective than aspirin in diabetic patients (83). However, larger-scale clinical trials are needed to substantiate these findings.
A highly selective inverse agonist of the 5-HT2A receptor has also been described (84). This agent, APD791, has successfully undergone phase 1 clinical studies.
Future Prospects for Developing Anti-Platelet Drugs
Other strategies for suppression of atherothrombosis are at an earlier stage from the perspective of drug development. Such strategies encompass targeting of the primary adhesion receptors on the platelet and of intracellular signaling pathways.
Glycoprotein Ib–IX–V
The first stage in the adhesion of platelets to subendothelial elements that are exposed by rupture of an atherosclerotic plaque involves a transient interaction between von Willebrand factor (VWF) and the glycoprotein Ib–IX–V complex (GpIb–IX–V), its primary receptor on the platelet (2, 3). Agents that block this interaction thus have considerable potential as anti-thrombotic drugs (85).
A number of strategies have been devised to effect such blockade (85–87). One approach has involved use of a recombinant fragment of the VWF A1 domain, amino-acid residues Leu1268—Lys1491, to compete for interaction with GpIb–IX–V (88). Although this approach appeared successful in animal studies, it has not led to a useful therapeutic agent. Other strategies have utilized a humanized monoclonal VWF-specific antibody, AJW200, or a humanized VWF-specific nanobody (i.e., a therapeutic molecule composed only of one variable domain from the heavy chain of an antibody), ALX-0081, to block the interaction (89, 90). Each agent has appeared to be safe and effective in initial phase 1 trials (90, 91). The most promising strategy to date, however, involves ARC1779, a nuclease-resistant aptamer that binds selectively to the activated A1 domain of VWF (92). Small-scale clinical trials have documented the safety of this aptamer and shown that it can raise platelet counts in patients with thrombotic thrombocytopenic purpura (91, 93). As this disease results from the presence of ultra-large VWF multimers that promote thrombosis, the therapeutic effectiveness of ARC1779 suggests that it will also be effective at suppressing atherothrombosis. The interaction of VWF with platelets is a particularly attractive target, in many respects, because it plays a critical role in platelet activation in the high shear environment of an atherosclerotic artery, yet only a minor role under conditions of lower shear stress (2, 3).
Collagen and Its Receptors
Interaction with subendothelial collagens also contributes substantially to platelet adhesion and activation when a section of atherosclerotic plaque ruptures (3, 94). Collagen plays an indirect role through binding of VWF and facilitating its interaction with platelet GpIb–IX–V. Collagen also interacts directly with two distinct receptors on the platelet, glycoprotein Ia–IIa (GpIa–IIa, integrin α2β1) and glycoprotein VI (GpVI). The GpIa–IIa receptor is expressed on a variety of different cell types, whereas GpVI is restricted to the platelet and is the preferred target for an anti-platelet drug. A number of inhibitors of collagen interaction with the platelet have been identified and examined in pre-clinical studies (41, 86, 95). Most progress toward therapeutic use has been made for PR-15, a humanized fusion protein that incorporates the extracellular domain of GpVI (96). It inhibits adhesion of platelets to exposed subendothelial collagen by competing with the GpVI receptor on the platelet. The safety of PR-15 has been demonstrated in a phase 1 trial (97).
Intracellular Signaling Pathways
The intracellular signaling pathways that lead to platelet activation and aggregation are also possible targets for antithrombotic drugs. Aspirin essentially targets one such pathway. As more knowledge has been obtained about the intricacies of intracellular signaling mechanisms in the platelet, more opportunities to inhibit such signaling have arisen.
A particularly appealing strategy is the suppression of intracellular signals that occur in the platelet when VWF engages the GpIb–IX–V receptor under conditions of high shear stress. This should theoretically provide selectivity for inhibition of atherothrombosis relative to hemostasis. It may thus lead to an antithrombotic drug with less tendency to cause bleeding. Pre-clinical studies have suggested that the p110β isozyme of Type Ia phosphoinositide 3-kinase participates in sustaining the activation of GpIIb–IIIa required for platelet aggregation under high shear conditions (98). Selective inhibitors of this isozyme, such as TGX-221 and CBL1309, appear to offer promise for the development of novel drugs to treat cardiovascular disease (98, 99). Other signaling pathways downstream of GpIb–IX–V may also offer invaluable targets for new antithrombotic drugs.
Conclusions
The current standard of care for patients with atherothrombotic diseases involves dual anti-platelet therapy with aspirin and clopidogrel. This strategy has substantially improved the therapeutic outcome, but it has significant limitations: recurrence of adverse vascular events remains a problem in some patients, whereas bleeding occurs in others. These limitations have provided a major impetus for developing new anti-platelet drugs. Prasugrel is an improvement over clopidogrel in terms of the efficacy and consistency of P2Y12 receptor blockade, but this is counterbalanced by an increased risk of bleeding. Clinical trials of other P2Y12 receptor antagonists and drugs directed against novel platelet targets have yielded hints that some of these avenues for drug development may yield agents with greater antithrombotic efficacy without an increased risk of bleeding. This is thus an exciting time in terms of prospects for better anti-platelet drugs.
Acknowledgments
We apologize to the many authors whose work has not been discussed due to constraints of space. This work was supported in part by Grant-in-Aid 0855297E from the Greater Southeast Affiliate of the American Heart Association (to JCK).
Footnotes
-
Authorship contributions
Wrote or contributed to the writing of the manuscript: Choi and Kermode.
- Copyright © 2011
References
Jaehwa Choi, PhD, is Assistant Professor of Pharmaceutical Sciences in the College of Pharmacy at Southwestern Oklahoma State University. She earned her doctoral degree from the University of Mississippi Medical Center. Her research interests focus on signal transduction mechanisms in cardiovascular disease and cancer.

John C. Kermode, PhD, is Professor of Pharmaceutical Sciences in the School of Pharmacy at the Philadelphia College of Osteopathic Medicine–Georgia Campus. He earned his doctoral degree from the University of London in England. His research interests are in the area of signal transduction mechanisms in the platelet with particular reference to von Willebrand factor. Send correspondence to JCK: E-mail JohnKer{at}pcom.edu; fax 678-407-7347.


