Mind the dbGAP: The Application of Data Mining to Identify Biological Mechanisms
One of the greatest challenges for a basic scientist is identifying genes that contribute to the biological mechanisms relevant to mammalian development and disease. For several decades, scientists have adapted animal model systems to identify genes underlying diverse biological phenomena. Sequencing of the human genome and creation of the human genetic map has led to the development of fixed-content genotyping assays that test single nucleotide polymorphisms (SNPs) across the human genome (1). These large genotyping efforts have served as the cornerstone of a new form of unbiased gene identification screen: the genome-wide association study (GWAS). The identification of a large number of genes associated with human traits and diseases within the past decade demonstrates the tremendous power of GWAS to provide insight into human, and more broadly, mammalian biology and disease (2). Following successful gene identification and independent replication, the baton is passed to the basic science researchers working to reveal the molecular mechanism that underlies the association.
Often as a requirement of funding or publishing this type of expansive work, researchers have deposited their data in the database of Genotypes and Phenotypes (dbGAP), an open and ever-expanding repository that is accessible to the general scientific community (3). The availability of such a large amount of human genetic data in dbGAP has created opportunities for scientific discovery, but how can the basic science researcher discern important signals among the noise? This Viewpoint will discuss approaches to mining dbGAP data for the identification of genes relevant to mammalian and human development and disease.
Classical genetic studies founded on the analysis of well-characterized phenotypes and on genetically studied animal model systems have been used extensively to identify genes responsible for diverse biological processes. Drosophila melanogaster was one of the first model systems employed for genetic analysis of development. With the added benefit of being a vertebrate organism, the zebrafish (Danio rerio) has also served as an excellent model organism for mammalian development (4). Both Drosophila and zebrafish have been subjected to random mutagenesis screens (i.e., the mutations were introduced into the genome in a manner not biased by the investigator), and these studies have led to the identification of genes required for developmental and homeostatic pathways in these organisms. Technical constraints have largely prevented the application of such approaches (i.e., unbiased genetic mutagenesis) in mammals. Rather, investigators have sought to use naturally occurring gene mutations and polymorphisms to identify genes that underlie mammalian and human development and disease. By starting with humans rather than animal model systems, the investigation occurs directly within the larger context of human traits and diseases. Indeed, the success of gene identification through analysis of Mendelian disorders in humans clearly demonstrates the benefits of performing gene identification studies in humans. The genes that underlie over 1,000 Mendelian syndromes have been identified so far using a variety of analytical approaches (5). Such single-gene disorders, however, are unable to fully explain major population health issues such as obesity, high blood pressure, and sporadic cancers, each of which are thought to be multigenic in nature.
Twin and family studies provide strong support for a genetic basis underlying numerous complex human traits (6). The challenge has been to identify the gene(s) responsible for the human heritability component of traits and diseases (as distinct from environmental and epigenetic influences). Sequencing of the human genome revealed that the majority of human sequence variation exists in the form of single nucleotide base differences, called single nucleotide polymorphisms (SNPs) (7, 8). Variation is also present in the form of variable numbers of short repeated sequence elements called microsatellites (9) as well as other relatively large structural variants (10), though these account for a much smaller fraction of inherited variability compared to SNPs. Identification and annotation of the human genome has led to the creation of a map of human genetic variants. High-throughput genome sequencing is likely to produce even more texture and granularity to the existing map of human genome variation (11). The availability of a dense set of SNPs and microsatellites serving as the basis of the human genome map supports the analysis of genes that underlie the genetic basis of complex human traits and diseases.
With available SNPs and microsatellites in hand the original experimental protocol for candidate gene association studies was roughly as follows: the researcher formulated a hypothesis, obtained genetic material from a cohort or case control study, designed a custom genotype assay discriminating typically fewer than one hundred markers, examined (“interrogated”) the genetic material for mutations or polymorphisms, and then tested the association of the genetic variant with the phenotype. By this point, the researcher would have invested a significant amount of time, money, and resources, creating a strong bias toward publishing findings even of marginal statistical significance (12). In hindsight, we learned that candidate gene association studies were overly focused on a limited number of genes and markers, and the standards for significance testing were overly lax (13).
The limitations of candidate gene studies were largely circumvented by the availability of a dense human genome map and the subsequent development of high throughput genotyping assays performed on microarray platforms. Assays initially capable of measuring ten thousand loci gave way to those measuring hundreds of thousands, and now over one million genetic markers. The result of this rapid technological development has been the commercialization of fixed genotyping microarray platforms that can interrogate large numbers of SNPs located in high density across the genome (14, 15). These platforms are considered “fixed” because their content cannot be customized for a given project. Rather, each genetic marker is included because of its ability to identify a unique allele and because of its adaptability to technical concerns inherent to the platform itself, and not because of any a priori investigator hypothesis. Increasing the number of individual assays included in each platform, along with the development of computational methods of genotype prediction (called imputation) based upon patterns of linkage disequilibrium endemic to the genome and observed between SNPs within populations, has led to the ability to identify nearly all of the common alleles in the human genome (generally defined as those variants present in over ten percent of the general population). Application of fixed genotyping arrays to population cohorts as well as case-control groups assembled to study a specific disease or phenotype forms the basis of modern GWASs.
Remarkably, the experimental approach of unbiased GWASs has offered tremendous advantages over the hypothesis-driven candidate gene association approach (16). First, GWASs have confirmed a large number of genes already known to be associated with traits through earlier analysis of specific pathways, candidate-gene based studies, and bench research. For example, genes known to be critical regulators of lipoproteins were found to have sequence variants that were associated with lipoprotein concentrations in the blood (17). Second, GWASs have identified genes not previously known to be associated with a trait or with the underlying biological processes relevant to the trait of interest (18). For example, the lipoprotein GWAS cited above also identified genes not previously known to be associated with lipid metabolism that turned out to be strong candidate regulators of lipid transport. In this regard, the possibility of gaining “new knowledge” through unbiased interrogation of the genome is perhaps the greatest strength of GWAS: the potential to provide biologists and other scientists with unique insight into the underlying basis of complex diseases and traits (19). Finally, un-biased testing of the entire genome provides greater perspective into many earlier results founded on the premises and technical limitations of candidate gene association studies (20, 21).
Demonstrating an association of a genetic polymorphism with a trait indicates that the local region of the genome exists in more than one form (allele) and that one or more of the different forms either provides protection from or contribution to that trait. The association of an SNP with trait is founded on the principle that the trait-associated variant is in linkage disequilibrium (LD) (Box 1) with one or more genetic mechanisms responsible for the biological effect. For example, the association may be founded on differences in gene expression caused by altered promoter or enhancer elements. Alternatively, the interrogated SNP may be in linkage disequilibrium with a variant that causes alternative exon usage or that alters the amino-acid sequence of an expressed peptide. Gene variants that alter the amino-acid sequence may significantly affect the biological function of the peptide. Recently, even coding variants that do not alter the peptide sequence have been identified as having an impact on cellular function through preferential codon usage (22). In any of these cases, finding a genetic association is the beginning of experimental work required to determine the underlying biological mechanism that forms the basis of the association.
Linkage Disequilibrium (LD)
Two SNPs associated in a nonrandom manner are considered to be in LD. Practically, this means that when there is complete LD between two SNPs, the genotype of one SNP can predict the genotype of the other SNP. By comparison, SNPs that are in linkage equilibrium are randomly associated, and the genotype of one SNP cannot predict the genotype of the other SNP. Degrees of LD are reported by the D′ and the regression coefficient (r2), both values range from zero to one. A D′=1.0 and an r2 =1.0 indicates complete LD while an r2 > 0.8 but less than 1.0 is consistent with near complete or partial LD. Multiple SNPs in LD form a haplotype block, which can extend for thousands of bases. Haplotype block size can be different between major human racial and ethnic groups.
The principle of LD critically underlies GWAS because SNPs chosen to be included on fixed genotype panels are not likely to be functional, which is to say they are unlikely to be the cause of or contribute directly to the trait under study. However, SNPs are included on a fixed genotype panel because if they may be in LD with variants actually responsible for the trait. When a GWAS identifies a SNP association with a trait, follow-up studies are performed to interrogate all variants in complete or partial LD with the GWAS SNP to identify the variant(s) that may be directly responsible for the trait association.
Many investigators have wondered why possibly causative SNPs in a particular gene—known from mouse or other animal model studies to have a critical role in a trait—have not been identified by GWASs. The investigator may not know with appreciable confidence why a negative association finding occurred, but whatever the underlying experimental design may be, it is important to remember the limitations of GWAS (23). First, genes can exist in which no variants have been identified—referred to as “monomorphic” genes—and, therefore, do not have a variant form that can be associated with a trait (24). Human evolution may have prevented the emergence of variant forms of genes critically required for development and maintenance of the species in times of selection pressure, thus creating such a functionally monomorphic locus in a population (25–27). Second, many older, fixed genotyping platforms had gaps in allele coverage or suffered from incomplete allele sampling. Provided the role of a particular gene in a trait has not been overestimated from animal or in vitro studies (12), GWAS should not be looked upon as necessarily excluding a role for a gene in a trait; a denser analysis of variants in the region may ultimately uncover the expected association.
Another concern relative to the long-term utility of GWAS is the persistent observation of apparently missing heritability (28, 29). That is, even though hundreds of heritable disorders have been associated with particular variants, the individual and cumulative effect sizes of these genes, traits, and associations have proven to account for only a small fraction of the total heritability of the trait estimated prior to the study. One possibility is that LD blocks tend to dilute the observation of any individual causative variant located within them (30). As a result, single, rare variants of large effect within populations exist alongside experimentally detected SNPs of lesser effect size. This type of locus has been observed in several Mendelian dyslipidemias (31, 32). Massive sequencing studies aimed at specific intracellular signaling pathways or disorders, such as one conducted in ANGPTL4, have aimed to collect systematically all variants in thousands of individuals. The resulting collection of variants, many previously unknown, does begin to account for a sizeable fraction of the “missing” effect size (33). The interaction of two or more genes affecting the same trait may also account for missing heritability (34, 35). Likewise, variable DNA methylation and shared environment can affect heritability estimates (36, 37).
How do we mine the data in the dbGAP? With the success of GWAS overcoming the key limitations of candidate gene–association studies and as a robust approach to identify disease- and trait-associated gene variants, a question naturally follows: can additional information be derived from GWAS datasets beyond the primary published results?
Recognizing the importance of the new genetic data produced through GWAS, the NIH and the extramural scientific community have worked together to produce dbGAP (3). The same model for providing the scientific community access to genome-wide genotype data is also being applied to the results of next-generation genome sequencing studies as they are completed (11, 38). The net effect of these initiatives is the availability of a large amount of human genotype data to the scientific community, which will help inform the design of future studies. The most remarkable effect is a change in the process and speed with which a genetic hypothesis may be tested. It is now possible to obtain genetic data from dbGAP and perform in silico association analyses without the burden of acquiring and analyzing any genetic material. Furthermore, because many cohorts have used identical or highly comparable genotype platforms, in silico meta analyses are also possible, increasing the power of detection by creating large cohorts from many smaller studies (39–41). The efficient analysis of existing genotype data from dbGAP holds the promise to conserve important DNA stocks while saving money and allowing for riskier hypotheses to be tested than would be practical under a candidate gene model. For example, it is always difficult to predict whether a given trait has a small number of genetic contributors each with a strong effect, or whether a trait was supported by a very large number of gene variants each with small individual effects (6, 16, 42). Mining dbGAP data may help predict the likelihood that a strong genetic basis underlies a given trait before time and money are spent in the collection, processing, and analysis of even a small, exploratory dataset.
It is supposed that the availability of extensive genetic data from thoroughly annotated phenotypes places tremendous discovery opportunities at the disposal of the scientific community. Although the advent of dbGAP has created tremendous excitement for the mining of large databases, the enthusiasm is tempered by the requirements of handling and analyzing these datasets. From our experience, one of the first challenges upon accessing such data is the sheer volume of material that must be organized and housed in a manner that protects the research subjects and adheres to the research mission. For example, the Framingham Heart Study dbGAP dataset includes thousands of individual phenotypic variables coupled to 549,915 genotypes from 9,274 individuals in > 1,000 families spread across three generational cohorts and two consent groups (43). A sophisticated data management approach is required to unpack, organize, and analyze such a large amount of interrelated information in any scientifically meaningful way. As always, prior preparation is key to mining fully such a deeply informative dataset. Highly focused research into a single phenotype of interest is possible with less preparation of data tables; however, that approach may miss important opportunities for discovery that crosscut seemingly unrelated phenotypes. Although the Framingham Heart Study has grown to encompass a multitude of observed phenotypes, many of the other datasets available through dbGAP represent highly focused cohorts that target single diseases (e.g., schizophrenia or prostate cancer).
The availability of nonsynonymous coding variant data contained in dbGAP datasets is one potential area for significant impact for molecular and cellular biology researchers. There are several reasons to consider selective analysis of nonsynonymous coding variants from dbGAP datasets outside of the context of a full GWAS. Variants that alter the peptide sequence have a significant ability to directly affect the biophysical properties of a protein and by extension to exert a cellular phenotype. Partnering the analysis of such coding variants with in vitro cell culture models and, ultimately, genetic association studies may provide significant new knowledge of a gene’s function.
Researchers employing classical molecular and cellular biology approaches may be unaware of the availability of data on naturally occurring variants contained within fixed genotype platforms in genes that they study. Newer fixed-content genotype panels are enriched for coding variants offering an even greater opportunity for gene-based discovery. The web-based bioinformatics tool SNAP (SNP Annotation and Proxy search) offers a way to query the content of fixed genotype panels for nonsynonymous variants (44). If the nonsynonymous SNP is not directly included on a fixed genotype array platform, a proxy SNP may be identified instead, based on observed complete or near complete linkage disequilibrium between the nonsynonymous SNP and the proxy. With SNAP, it is possible to return every proxy present (for a nonsynonymous SNP) within the HapMap or 1000 Genomes datasets at a preferred level of confidence and then automatically filter the results relative to available genotyping platform. Alternatively, SNAP will simply return all known variants within the region, which might be useful in performing a directed genotype association analysis within a cohort across an entire region of LD. In this way, excellent proxies for non-synonymous variants can be quickly identified, vastly improving the scientist’s capability for fast and convenient independent replication analysis using preexisting genotype data from dbGAP.
Despite the conceptual strengths of prioritizing the analysis of nonsynonymous variants from dbGAP, several limitations must be acknowledged. Functional coding variants often have a low minor allele frequency, which introduces several potential problems in genetic association studies. Fixed GWAS platforms, which are founded on the common-gene common-disease hypothesis, typically exclude rare coding variants because of a reduced power to detect an association when the observed minor allele frequency is less than ten percent. Differences in rare allele frequency between racial and ethnic groups particularly confound association analyses (45). Finally, variants with an allele frequency below one percent are subject to significant artifact introduced by measurement error. Family-based studies have a particularly important role in rare variant analysis. Demonstrating transmission of a rare variant within a family alleviates concerns about genotype error and population stratification and greatly improves the ability to analyze the effects of rare variants even when restricted to a few families in a large cohort.
Strict correction for multiple hypothesis testing with the Bonferroni technique (Box 2) has helped focus GWAS results on gene variants that have durable associations with phenotype in part by rendering many SNP-phenotype associations below the level of significance. Correction for the effects of multiple hypothesis testing is required to reduce the noise in the assay results at the expense of eliminating many important variants from consideration (20, 21, 46, 47). The current approach to identifying new variants that have important trait associations is to simply increase the number of subjects tested by combining cohorts. However, increasing the number of subjects for analysis is not always possible for rare phenotypes. One approach to discovering trait-associated variants is to relax the threshold for significance. This approach is supported by the fact that fixed-genotype genome wide panels contain multiple variants in near or complete LD and often have a gene-centric variant density pattern. Because such variants in partial linkage disequilibrium are not fully independent, correcting for every single gene variant tested would seem to be overly conservative. Indiscriminately reducing the level of correction for all markers would increase the number of gene variant associations considered to achieve statistical significance at the expense of increasing the number of false associations. Another approach is to apply a correction based on LD patterns and marker density; this approach may achieve a more accurate correction threshold for significance testing (48).
Bonferroni Correction and Multiple Testing Artifact
Many scientific studies testing a single hypothesis specify in advance that the results must surpass a threshold for significance, stating that if the finding could have occurred by chance less than five percent of the time (denoted by p<0.05), then a significant discovery will be concluded. Each genotype association is a separate hypothesis, which means that within each GWAS hundreds of thousands if not millions of hypotheses are tested at one time. The large number of hypotheses being tested therefore means that if a five percent cut-off were used, then a very large number of results will surpass the pre-specified threshold by random chance alone. Multiple hypothesis testing in GWAS is therefore more likely to identify false associations than true associations when the p<0.05 threshold is used.
There are many approaches to correct for multiple hypothesis-testing artifacts with the goal of reducing the number of false associations. The most conservative approach is the Bonferroni correction, which establishes the threshold for significance at a p value less than 0.05 divided by the number of hypotheses being tested. Said another way, the experimental p value multiplied by the number of hypotheses tested must be less than 0.05 (or whatever threshold for significance is chosen a priori). For example, because all observations are considered independent by this correction model, a 500,000 genotype assay will require the p value for association to be lower than 0.0000001 (i.e., 0.05/500,000) to be considered likely not the result of chance alone. It is the implicit consideration that all genotypes on fixed genotype platforms are fully independent (when in fact many are in LD with each other) that has lead to the Bonferroni correction being widely considered “overly conservative.” However, no compelling replacement that addresses the very real problem of false associations in GWASs has been widely adopted.
Our approach to reduce the burden of multiple test correction is to continue to use the Bonferroni correction protocol but to reduce the number of genotypes tested against a trait by selecting from the fixed-content genotype panel only those markers likely to be informative relative to the trait under study. These markers include those contained within genes relevant to the underlying biological process under study. Selection of all genes relevant to a trait in an unbiased and as comprehensive a manner possible, using all available information, is the critical aspect that differentiates the pathway approach from candidate gene studies. Gene selection is accomplished by employing computer programs to mine the published literature (Figure 1). Genes with an altered expression pattern (as measured by gene expression microarray) within a trait or a targeted tissue type are added to the list. Finally, genomic loci previously associated with traits can be added to the gene selection algorithms. Once a list of genes and loci are compiled, they are interrelated through the application of gene pathway programs, such as STRING, CANDID, or Endeavour, that establish both links between input genes and well-known interactions that may not have been included in the input (49–51). Importantly, these programs identify functional interactions between gene products and frequently offer a rank order of importance of genes based upon their level of interrelatedness. Genes that serve as nodal points that interact or function with multiple different genes tend to have the greatest importance, as defects within these genes could potentially have broad ripple effects throughout the entire pathway. Network information in hand, genes and their SNPs contained within fixed genotyping platforms can then be subdivided and interrogated against the trait under study with a high probability of being informative relative to a network of interest.
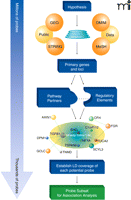
Pathway analysis tools can use biological knowledge to focus the number of genetic markers for association analysis from millions of potential variants to a few thousand tightly focused on the trait under study. The initial hypothesis, based upon a known gene and/or a biological process, forms the basis for selecting Medical Subject Headings (MeSH), which are used to mine both the published literature and the Online Mendelian Inheritance in Man (OMIM) databases. This list is supplemented with genes that are differentially expressed in the setting of the tissue or trait of interest identified from analysis of Gene Expression Omnibus (GEO) microarray datasets. The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) facilitates this process by identifying likely pathway-related genes based on a wide array of knowledge-based interrelationships, including protein interaction, known co-regulation, and comparative genomics. These data-mining steps serve to generate a primary list of genes deemed biologically relevant to the trait under study. Analysis of pathway relationships further builds out the gene list as well as stratifies the gene list by identifying molecular relationships and key partners. All genetic markers present on fixed genotype platforms that are also contained within or nearby pathway genes (e.g., in upstream or downstream regulatory sequences) are then identified. Pathway SNPs that provide similar overall information content by virtue of being in high LD are removed to focus the list of markers and reduce the overall testing penalty. Finally, the refined list of Pathway SNPs is used to test genotype-phenotype associations in novel or dbGAP-derived datasets. ENG, endoglin; TGFB1, transforming growth factor–beta 1; DPM1, dolichyl-phosphate mannosyltransferase polypeptide 1; GCLC, glutamate-cysteine ligase; TNMD, tenomodulin; TSPAN6, tetraspanin 6; SCYL3, SCY1-like 3; FUCA2, fucosidase, alpha-L- 2; FGR, Gardner-Rasheed feline sarcoma viral (v-fgr) oncogene homolog; CFH, complement factor H; AXIN1, axin 1; C1orf112, chromosome 1 open reading frame 112; NFYA, nuclear transcription factor Y, alpha.
Networks based on biological knowledge, even when extended beyond “key” genes to their partners and regulators, still rely upon the existing knowledge base. It is therefore desirable to create a method by which new information can be derived through the analysis of SNPs likely to have information content relative to the cohort. This approach is broadly referred to as Random Forests (52–54). Briefly, genotypes of the entire platform are randomly divided into a test and training groups; these groups are then repeatedly permuted versus trait to find SNPs that appear to have important information content because of their ability to subdivide the trait efficiently. The random division process itself is repeated and re-permuted. Ultimately, a list of SNPs can be generated that likely have information content. These “important” SNPs can then be analyzed versus trait directly with no multiple testing penalty relative to the permutation step. Technical limitations relative to the ability to permute large genotyping platforms sufficiently to fully model all possible combinations and thus truly extract the information content of the ideal subset of SNPs have thus far limited this approach to smaller platforms. Recent advances, however, in a related approach, Random Jungle, hold the promise of advancing these sorts of fundamentally ab initio probe selection approaches into the realm of the bench scientist (54).
In our experience, the application of the pathway-based approach offers a tenfold reduction in multiple testing burdens (18). The application of pathway-based tools for the selection of variants for analysis offers a reasonable opportunity to identify trait-associated variants that do not surpass full Bonferroni correction of multiple hypothesis testing without sacrificing the importance of a priori biological knowledge.
The availability of genome-wide genetic data from large well-phenotyped cohorts and case control studies offers an unparalleled opportunity to understand and to research the genetic bases of human traits and diseases in humans rather than animal models. GWASs have already demonstrated important new genes whose role in disease has been confirmed and which are currently under investigation for potential therapeutic development. Beyond the primary results from GWAS, there are opportunities for both population and bench scientists alike to make new discoveries using archived dbGAP data. Scientists who devote their work to a particular gene or set of genes may find phenotype associations that will direct their research in new and unexpected directions. Training scientists to have the necessary skills, providing suitable infrastructure to effectively mine the dbGAP data, and maintaining high ethical standards toward handling these data are an important part of realizing the potential of this tremendous repository of human genetic information.
Acknowledgments
This work was supported by the National Institutes of Health [Grant HL077378] and the American Heart Association [Grant 0816005D] (E.C.W).
Footnotes
-
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Huggins and Wooten.
- Copyright © 2011
References
Eric C. Wooten, PhD, is an Instructor in Medicine at Tufts University and Research Associate at Tufts Medical Center in Boston, MA. He received his degree in molecular and cellular biology from Baylor College of Medicine in Houston, TX, and was subsequently a postdoctoral fellow at Boston University Medical Center and, later, at the Molecular Cardiology Research Institute (MCRI) Center for Translational Genetics at Tufts Medical Center. His primary research interests are in human genetics and specifically genomic sequence, organization, and structure, and the repercussions that inherited and novel alterations to the genome have on the broader epigenetic, transcriptional, and molecular regulatory functions both in cells and within physiological systems. E-mail ewooten{at}tuftsmedicalcenter.org; fax 617-636-8692.

Gordon S. Huggins, MD, is an Associate Professor at Tufts University School of Medicine and an Investigator at the Molecular Cardiology Research Institute (MCRI) and Cardiology Division of Tufts Medical Center, Boston MA. Dr. Huggins directs the MCRI Center for Translational Genomics whose primary goal is to use human genetic variation and gene expression to investigate mechanisms that underlie human cardiovascular development and disease. E-mail ghuggins{at}tuftsmedicalcenter.org; fax 617-636-8692.


