Expression Array Technology in the Diagnosis and Treatment of Breast Cancer
- 1Departments of Surgery and
- 2Genetics Stanford University School of Medicine Stanford CA 94305 and
- 3Department of Genetics Norwegian Radium Hospital University of Oslo, Oslo, Norway.
- SSJ. E-mail ssj{at}stanford.edu; fax 650-498-6250.
Abstract
The most common group of cancers among American women involves malignancies of the breast. Breast cancer is a complex disease, involving several different types of tissues and specific cells with various functions, that is categorized into many distinct subtypes. Microarray analysis has recently revealed that different biological subtypes of breast cancer are accompanied by differences in their specific gene expression profile. Because breast tissue (and breast cancer) is heterogeneous, microarray analysis may provide clinicians with a better understanding of how to treat each specific case. Thus, microarray analysis may translate basic research data into more confident diagnoses, specifically designed treatment regimens geared to each patient's needs, and better clinical prognoses.
INTRODUCTION
Breast cancer is the most common malignancy and the second most deadly cancer in American women. It is a complex disease; there are multiple pathologic subtypes, and even among pathologically similar subtypes, clinical presentations and outcomes can be disparate. The two most common subtypes of breast cancer are infiltrating ductal carcinoma and infiltrating lobular carcinoma, which differ markedly both in the histology of the malignant epithelial cells and in the clinical course of disease. Among infiltrating ductal carcinomas, by far the most common pathologic subtype of breast cancer, there are three histologic grades that designate how similarly (“well-differentiated”) or dissimilarly (“poorly differentiated”) the cancer cells resemble normal breast epithelial cells by defined microscopic criteria. In the case of infiltrating ductal carcinoma, the poorer the resemblance, the poorer the clinical outcome, signifying a more aggressive form of breast cancer.
It is generally believed that invasive breast cancer derives from precancerous changes (atypical hyperplasia) that beget ductal carcinoma in situ (DCIS), also known as intraductal carcinoma, because the malignant epithelial cells remain confined within the basement membrane of the ductal tissue (1). Eventually, enough genetic changes accumulate so that these cells become able to pierce the basement membrane and invade the surrounding stromal tissue. In some cases, invasion is associated with a cascade of events that may include angiogenesis, lymphovascular invasion, lymph node metastases, and hematogenously conveyed distant metastases. Twenty-five to thirty percent of invasive breast cancers ultimately lead to the demise of the patient, in spite of our best efforts at treatment.
Recently, the powerful technology of microarray analysis has been exploited to explore gene expression in breast tissue on a genome-wide scale, and has shown that different biological subtypes of breast cancer are accompanied by differences in their transcriptional programs. With cDNA microarrays, the relative expression levels of tens of thousands of genes within a specific tissue sample can be measured simultaneously. Breast tissue is heterogeneous, with components of epithelial, mesenchymal, endothelial, and lymphopoietic derivation. Nevertheless, the influences of these cell types on the tumor's total pattern of gene expression can be estimated analytically. Thus, microarrays permit total tissue analysis and provide a strikingly stable molecular portrait of tumors.
MICROARRAY TECHNOLOGY
DNA microarrays owe their power as an experimental tool to the specificity and affinity of complementary base pairing. Many thousands of oligonucleotides or cDNA clones can be spotted onto a single glass slide microarray, and indeed most of the genome can now be interrogated in a single microarray. Thus, an expression level “snapshot” of cellular activity provides an unprecedented tool for exploring the behavior of the genome under almost any conditions of cell culture, and is amenable to repetitive analyses of frozen human tissue.
The Microarray Substrate
In principle, membrane-based arrays can be probed by radiolabeled mRNA to measure gene expression; however, slide-based arrays have proven to be smaller, more convenient, and to facilitate higher throughput. There are two important strategies in the fabrication of slide-based microarrays. One strategy is commonly referred to as “in situ oligo synthesis.” In this approach, pioneered by the Affymetrix® Corporation, sequences of fifteen to twenty-five nucleotides can be accurately and efficiently synthesized. In an alternative approach, developed at Rosetta Inpharmatics and Agilent Technologies, an inkjet printer, rather than photolithography, is used to apply sequential rounds of synthesis, using standard phosphoramidite chemistry, and allows the construction of oligonucleotides of sixty to eighty nucleotides in length. This latter style of in situ oligo array can permit competitive hybridization of two samples in the same manner as do the batch-synthesized arrays described below.
In the other major strategy, batches of “bio-ink” are synthesized in large quantities, and then printed on a substrate, usually a treated glass microscope slide (Figure 1⇓), through any of a variety of techniques, including both contact and inkjet printing. The bio-ink can be cDNAs from large expressed sequence tag (EST) libraries or long oligonucleotide chains produced in large-scale oligo synthesizers. Whereas in situ oligo arrays offer some flexibility in the choice and arrangement of features to be printed, bio-ink arrays benefit from the economy inherent in synthesizing reagents on a large scale.

Glass slide cDNA microarray.
One of the most popular techniques for printing is a simple contact printing technique (rather than inkjet printing as practiced, for example, by Agilent), developed by Pat Brown and colleagues at Stanford University. The Brown Lab style printers (http://cmgm.stanford.edu/pbrown/mguide/index.html) are relatively inexpensive and have attracted a following among academic laboratories and university core facilities. An investigator may use well over 100 microarrays in a well-designed experiment or in surveys of human tumor samples. Because of the large number of individual hybridizations performed in the course of an investigation, it is important to keep the cost of a microarray experiment as low as possible. At Stanford, the total cost (including hybridization reagents, reference RNA, and database and bioinformatics support) for a 42,000-spot array is about $200 per hybridization.
The IMAGE consortium and the National Cancer Institute's Cancer Genome Anatomy Project (CGAP) clone libraries are the source of the human cDNA microarray developed at Stanford. The IMAGE clones were obtained through Research Genetics Corporation, who manage the clone sets and verify sequences. Clones are selected in order to represent unique clusters of ESTs as defined by UniGene (http://www.ncbi.nlm.nih.gov/UniGene/). On a typical array produced at Stanford from these clone sets, 40% of the cDNAs will represent genes with a gene symbol in UniGene, 10% will represent ESTs with non-trivial annotation (i.e., the gene has been studied but has not yet been assigned a gene symbol), and 50% will represent ESTs with little or no annotation.
Probe Preparation
Either total RNA or mRNA can be isolated from snap-frozen tumors and used for microarray hybridization. Standard protocols require relatively large amounts of RNA (2–4 micrograms of mRNA or 50 micrograms of total RNA) to achieve adequate signal for weakly expressed transcripts. Because these amounts of RNA probes require larger starting quantities of human tissue than is often available, RNA amplification strategies have become important. These methods permit the use of much smaller quantities of total RNA while still maintaining excellent fidelity with respect to the original tissue sample. When arrays are used to explore the biology of individual tissue components (such as epithelial cells or stromal fibroblasts), laser capture microdissection can be used to purify cell populations (2).
The isolated RNA is labeled with fluorescent dyes. By convention, the experimental sample is labeled with Cy 5, a red dye that fluoresces at 635 nm, by means of a reverse transcription reaction. The Cy 5–labeled cDNA sample is mixed with a reference cDNA that has been labeled with Cy 3, a green dye that fluoresces at 532 nm, and the mixture is hybridized to the array (Figure 2⇓). An optical scanner measures fluorescence at the two specified wavelengths, and the ratio of signal intensities from the experimental and reference RNA represents the relative abundance of transcripts present in the sample. This use of ratios permits the canceling out of systematic errors, such as unknown quantities of DNA spotted onto the array and differing hybridization kinetics. Reference RNA, now commercially available, is composed from RNA pooled from ten cell lines that express the majority of human genes and provide a renewable resource. All expression levels in the sample are reported relative to the nominal level provided by the reference RNA, which permits samples using the same reference RNA to be compared (3–,5).
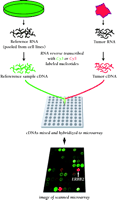
Schematic of microarray technique. RNA from a tumor sample and reference RNA (made commercially from pooled cell cultures to represent the majority of known genes) are reverse transcribed and labeled with different fluorescent dyes. The mixture is hybridized overnight to a microarray. The hybridized microarray is then scanned at two wavelengths and the intensities of red and green fluorescence are measured at each spot on the microarray. The red-to-green ratio reveals the abundance of RNA expressed by the tumor sample relative to the reference sample for every one of the 42,000 cDNA clones on the array. This technique provides a comparative measure of the global gene expression of the tumor sample.
Data Retrieval and Analysis
Because microarrays measure the transcription of genes on a genome-wide basis, they detail the cell's metabolic status in terms of which genes are induced or repressed. More importantly, such widespread expression data provide information on relationships among genes that may not have been previously suspected. When microarrays are used to study human cancer, they reveal information about the state of a tissue, even including interactions between malignant cells and their microenvironment. Examples of such interactions include tumor angiogenesis, which is an endothelial response to an epithelial malignancy, or the infiltration of tumor-associated lymphocytes. The complexity of interactions between different cell types in a tissue is thus measurable with microarrays; the analysis of such data, however, presents a formidable problem.
The output of experimental information from cDNA microarrays is a plethora of fluorescence intensities. These raw data must be stored in a retrievable format, analyzed, and optimally, subjected to a visualization method so that researchers can use their own intrinsic neural networks (i.e., their brains) to interpret correlations among numerous diverse but interrelated data points. Web-based formats such as the Stanford Microarray Database (SMD; http://genome-www5.stanford.edu/MicroArray/SMD) provide such a repository for data storage and analysis. SMD provides a Web-based link between specific data (gene spots) and publicly accessible databases, such as UniGene, dbEST, and Swiss-Prot, for valuable information about the clones used in array preparation (http://genome-www5.stanford.edu/cgi-bin/SMD/source/sourceSearch). Moreover, SMD is a resource for public dissemination of expression data for all published studies.
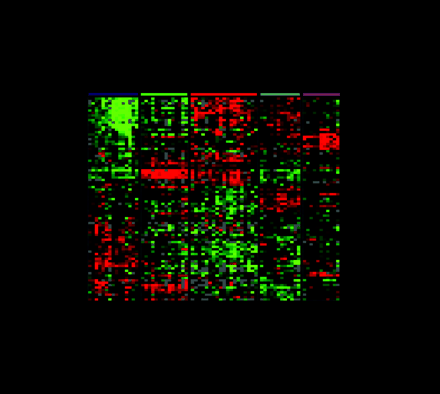
One of the most commonly used methods for microarray data analysis is hierarchical clustering (6), a method for organizing genes and experimental samples according to similarity of gene expression profile. In this method, a mathematical vector for the expression profile of each gene in a sample is determined and compared to the vectors from all of the genes expressed in that sample. Genes with similar expression patterns can then be clustered near one another along one axis, and similarly, experimental samples can be ordered according to their overall similarity in gene expression patterns along a second axis. The result is that genes whose expression patterns are most similar are clustered together by rows, and experimental samples whose expression patterns are most similar are clustered by columns (Figure 3⇓). Dendrograms linking genes or experimental samples can then be generated to show degrees of similarity. In this way, useful relationships between coexpressed genes or similar experimental samples may be discovered. Besides hierarchical clustering, algorithms that detect pattern similarity include k-means clustering and self-organizing maps. Singular value decomposition (principal component analysis) is used to identify patterns that contribute to the overall gene expression matrix (7). Neural networks may apply analytic techniques to a training set of data and test the result on a separate validation set of samples.
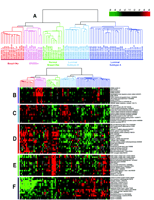
Gene expression patterns of eighty-five breast samples. Seventy-eight carcinomas, three benign tumors, and four normal breast tissues cluster into five subtypes: Luminal A [estrogen-receptor (ER) positive, favorable survival]; Luminal B (ER positive, poor survival); Normal breast-like; ERBB2 amplicon; Basal epithelial-like cluster.
(A) Tumor clusters are represented by branched dendrograms (upper figure) that indicate degree of similarity between samples. Genes are clustered by rows according to similarity of expression. Red indicates high gene expression relative to reference; green indicates more expression in reference RNA than in tumor sample (low relative expression). Representataive gene clusters expressed by the five tumor subtypes above are shown: (B) the ERBB2 amplicon cluster; (C) genes coexpressed by the Luminal B tumors and the basal and ERBB2 tumors; (D) basal epithelial cluster containing keratins 5 and 17; (E) normal breast-like cluster; and (F) Luminal A cluster containing ER-associated genes with lower relative expression of these genes by the Luminal B tumors. [From Sorlie et al., PNAS 98: 10869–10874. Copyright (2001) National Academy of Sciences, U.S.A.]
Hierarchical clustering that is performed on entire data sets without expectation or previous information regarding results is known as “unsupervised clustering,” and offers the virtue that no assumptions are made. Gene expression patterns can also be determined using supervised methods, provided that one has prior information about the genes or tissue samples so that predictions of clustering can be made. Information such as clinical outcome, pathologic subtype, or sample similarity may be used to “supervise” an analysis in order to generate a pertinent gene list that may then be tested against a new set of unknown samples.
MICROARRAY TECHNOLOGY AND BREAST CANCER
The combination of expressed and repressed genes within a tumor reflects the global state of the tumor, revealing information pertaining to cellular metabolic rates, proliferative status, and even molecular interactions between malignant epithelial cells and surrounding fibroblasts, adipocytes, endothelial cells, tumor-associated macrophages, or lymphocytes. Indeed, the gene expression profile of a tumor provides a unique molecular “portrait,” or “signature,” that can be correlated with clinical behavior and drug responsiveness. Identification of specific patterns of gene expression will undoubtedly improve tumor classification, prognosis, and treatment schemes.
In 1999, we showed that microarrays could be used to elaborate transcriptional profiles of human breast cancers (8). Moreover, genes coexpressed among distinct cell types within breast cancer tissue could be discerned as clusters representing the concerted expression of genes from proliferating malignant epithelial cells, B lymphocytes, and stromal cells. Using a hierarchical clustering algorithm, expression information was grouped computationally in a manner analogous to what a cell sorter might do mechanically. For example, human mammary epithelial cell cultures grown to senescence or manipulated in vitro by growth factors demonstrated that gene expression clusters can reflect proliferation-associated cell cycle gene sets, epidermal growth factor-responsive genes, and interferon-regulated genes [including genes in the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathway] that otherwise varied with respect to expression in thirteen distinct human breast cancers studied. Gene expression in specific cell types was confirmed on paraffin sections using immunohistochemical stains.
More recently, we reported a study of sixty-five human benign and malignant breast samples from forty-two patients—thirty-nine carcinomas, including twenty tumor pairs sampled before and after chemotherapy, and two cancers with corresponding lymph node metastases (9). Expression data from nineteen cell lines comprising B cells, T cells, macrophages, endothelial cells, and epithelial cells were also studied. Using an 8102-gene array and a refined pool of reference mRNA from eleven different cell lines that represented a greater majority of known human genes, cluster analyses were more informative. Specifically, expression clusters could be used to identify proliferating epithelial cells, endothelial cells, stromal fibroblasts, B cells, T cells, macrophages, and adipose-enriched normal breast, and included genes such as those encoding peroxisome proliferative activated receptor γ (PPARγ) and fatty acid binding protein 4. An estrogen-regulated gene cluster was also identified that indicated expression of the estrogen receptor (ER), GATA-binding protein 3, acyl-coenzyme A dehydrogenase, hepatocyte nuclear factor 3a, X-box binding protein 1, N-acetyltransferase 1, and the estrogen-regulated LIV-1 protein. The epithelial cells from tumors expressing this cluster stained for keratins 8 and 18, suggesting a luminal origin or differentiation pathway. In contrast to the ER-positive luminal cluster, there was also an ER-negative (“basal”) epithelial cell–associated cluster producing transcripts for keratins 5 and 17, laminins, integrin b-4, S100 calcium-binding protein A2, annexin A8, matrix metalloproteinase 14, and small inducible cytokine subfamily D. An ERBB2 cluster included genes encoding growth factor receptor–bound protein 7 (GRB7), steroidogenic acute regulatory protein related (MLN64), and tissue necrosis factor receptor–associated factor 4 (TRAF4), which is on the chromosome 17 amplicon along with the ERBB2 gene (10,,11).
As mentioned above, our tumor set included two tumors with associated metastatic lymph nodes, and twenty late-stage breast cancers from Norwegian patients who underwent tumor biopsy, sixteen weeks of doxorubicin treatment, and then tumor rebiopsy and/or surgery. Most of these patients were also placed on longterm tamoxifen treatment following surgery. A very important finding of the study was that almost all the tumor pairs—the pre- and postchemotherapy Norwegian tumors, as well as tumors with corresponding lymph node metastases—showed such consistent similarity in gene expression that they almost always were paired together within the overall cluster, regardless of tumor subtype (luminal, basal, or ERBB2-expressing). This pairing occurred notwithstanding the fact that the tumor pairs were biopsied almost four months apart following intensive chemotherapy. Of the five exceptions, three were tumors that were responsive to preoperative chemotherapy, leaving residual breast tissue that clustered with genes expressed by “normal” nonmalignant breast tissue samples.
In a follow-up study (12), a set of seventy-eight human breast cancers, three fibroadenomas, and four samples of normal breast tissue were analyzed. Cluster analysis confirmed the previously defined of ER-positive luminal, ER-negative basal, ERBB2-overexpressing, and normal breast subgroups; however, in this expanded series, the luminal ER-positive tumors subdivided into two distinct subtypes (see Figure 3⇑). Luminal A tumors showed high expression of estrogen-regulated and associated genes as mentioned above. Luminal B tumors, although still ER positive, expressed lower levels of the genes associated with the ER cluster and also expressed some genes that had previously clustered with some of the ERBB2-overexpressing and basal tumors. The ERBB2-overexpressing and basal tumors (70–80%) tended to harbor mutations of the TP53 tumor suppressor gene, whereas 67% of the Luminal B group had and only 13% of the Luminal A group contained TP53 mutations. Moreover, clinical survival of patients with Luminal A tumors was markedly better than patients with tumors of other subtypes, in spite of the fact that the tumors in all groups were locally advanced, most larger than 5 cm with lymph node metastases. The prolonged and unexpected survival of patients molecularly subgrouped by their Luminal A tumors suggests either very favorable tumor biology (limited metastatic potential) or excellent responsiveness to doxorubicin and tamoxifen treatment. The Luminal B tumors, although also ER positive, were more deleterious; these tumors, in contrast to the Luminal A ER-positive subtype, produced transcripts encoding the myeloblastosis viral oncogene homolog MYB, gamma-glutamyl hydrolase (GGH), and other enzymes involved in cell signaling and sterol biosynthesis that distinguished them from the longer surviving.
Other correlations between (13-16) gene expression in breast cancer and ER status have been made (Tables 1 and 2⇓⇓). Many of the same genes that involved in the Luminal A subtype (see above) are coexpressed with the ER and confer a favorable long-term prognosis. Conversely, ER-negative tumors are characterized by different sets of genes and are associated with a less favorable prognosis. These differences in molecular phenotype may originate in precursor lesions, prior to the development of invasive breast cancer, that set the course for differences in tumor subtype (17). van't Veer and colleagues (15) examined expression signatures of ninety-eight node-negative breast cancers in women under fifty-five years of age, including seventy-eight sporadic cancers and twenty from women with germline mutations in the breast cancer susceptibility genes BRCA1 orBRCA2. Although most breast cancers generally occur in women after menopause, younger patients tend to present with more aggressive phenotypes, as is evidenced (15) by high tumor grade in over two-thirds of the patients and angioinvasion in nearly one-third. A list of seventy optimal marker genes has been developed that may predict prognosis in high-risk younger women (15). Ahr and colleagues (18,,19) have identified a different set of marker genes in fifty-five patients, a subgroup of whom shows high rates of cancer recurrence in short-interval follow-up. In contrast, Unger et al. (20) have reported on the expression profiles of two distinct, synchronous breast cancers, in a single breast, that had almost identical gene expression profiles significantly different from the expression profiles of tumors from other patients. This may suggest intrinsic patient-related factors that affect the course of malignant transformation. These collective data not only reflect the diversity of molecular phenotypes of breast cancer among patient populations, but also underscore, with the impending burst in groups that perform microarray technology, the urgent need for standardization of data across various platforms.
Gene Expression Associated with Estrogen Receptor- Positive Tumorsa
Correlation with Estrogen Receptor-Negative Tumors a
Microarrays are also being used to distinguish expression signatures of tumors with BRCA1 or BRCA2 mutations and to refine our understanding of these genes (15,21,,22). In one study, BRCA1-associated cancers are distinct from BRCA2-associated cancers in expressing genes associated with cellular responses to stress, such as DNA repair and pro-apoptosis genes, but tending to manifest low levels of expression for anti-apoptotic genes. BRCA1 tumors also tend to be enriched in infiltrating lymphocytes, concordant with the particular pathology of these tumors (23). The divergence of the two genotypes requires resolution before the results can be utilized for drug design and clinical care.
FUTURE HOPE
The use of microarrays to subclassify individual breast cancers for prediction of clinical outcome and response to systemic therapy is just now burgeoning. It is expected that within the next five years, analyses using microarray technology, in conjunction with information provided by single nucleotide polymorphism arrays and the growing field of proteomics, will introduce new sets of markers that may identify patients with a particular need for systemic therapy. Care and appropriate time must be taken with these trials, because slower growing, more differentiated tumors may take as long as eight to eighteen years before recurrence becomes clinically apparent. Metastases in axillary lymph nodes are currently our strongest predictor of clinical outcome. It is our hope that molecular analyses may eventually relegate lymph node biopsy and transition axillary lymph node dissection to therapeutic rather than diagnostic modalities. Moreover, for patients who do require systemic treatment, novel targeted therapies may be developed based on expression patterns that will individualize therapy in a tumor-specific and potentially less toxic manner, which is the promise of pharmacogenomics (24). Finally, using microarrays to study normal and precursor lesions, as can be done using laser capture microdissection (25), should help us answer some basic biological questions regarding malignant transformation and disease progression.
The application of microarray technology to breast cancer has already produced exciting and encouraging data. The goal now is to offer targeted therapies to those that require them—specifically to women with occult micrometastatic disease at the time of breast cancer diagnosis—and conversely, to prevent unnecessary toxic and expensive systemic therapies for patients with non-metastasizing tumors that are cured by excision alone. Identification of “at risk” women may offer hope for directing screening efforts selectively toward women in the population who will benefit most. And finally, as we begin to learn more about the earliest steps in malignant transformation, we may be able to devise methods for preventing or arresting this process. The future of genomics and its younger sibling, proteomics, promises to change the way we practice medicine.
Acknowledgments
We are grateful to the National Cancer Institute, the Norwegian Research Council, and the Norwegian Cancer Society for support for our research. We also wish to thank all the women who generously provided tissue for our studies.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Stefanie S. Jeffrey, M.D., (top left) is Associate Professor of Surgery at the Stanford University School of Medicine. Michael J. Fero, Ph.D., (top right) is Director of the Stanford Functional Genomics Facility at the Stanford University School of Medicine. Anne-Lise Børresen-Dale, M.D.,(bottom left) is the chair of Department of Genetics at the Institute for Cancer Research, Norwegian Radium Hospital, at the University of Oslo, Oslo, Norway. David Botstein, Ph.D., (bottom right) is the Stanley Acherman Professor in the Department of Genetics at the Stanford University School of Medicine.

