LuXuRies of Lipid Homeostasis: The Unity of Nuclear Hormone Receptors, Transcription Regulation, and Cholesterol Sensing
- Department of Pharmacology Howard Hughes Medical Institute University of Texas Southwestern Medical Center 5323 Harry Hines Blvd. Dallas Tx 75390-9050
- DJM. E-mail davo.mango{at}utsouthwestern.edu; fax 214-648-5419.
Abstract
Cholesterol homeostasis is maintained by a regulatory network that controls both the acquisition and elimination of cholesterol. Recent studies have elucidated a mechanism by which cholesterol metabolism is transcriptionally regulated by several classes of orphan nuclear receptors. In particular, the liver X receptors, LXRα and LXRβ, appear to serve as key sensors of intracellular sterol levels by regulating the expression of genes that control cholesterol absorption, storage, transport, and elimination. LXRs are also involved in fatty acid metabolism by their ability to increase the expression of sterol regulatory element–binding protein 1c (SREBP-1c). These findings define LXRs as potential therapeutic targets for the treatment of lipid disorders.
INTRODUCTION
Liver X receptors (LXRs) α and β are members of the superfamily of nuclear hormone receptor proteins (1). They are so named because a human LXRα that was first identified by screening a liver cDNA library is highly expressed in liver (2,,3). Like other nuclear hormone receptor proteins, LXRs are composed of a highly conserved zinc-finger DNA-binding domain and a less conserved ligand-binding domain. LXRα (NR1H3, also called RLD-1) and LXRβ (NR1H2, also called UR, NER1, OR-1, and RIP15) are highly related and share ~77% amino acid sequence identity in both their DNA- and ligand-binding domains (4). LXRα-encoding mRNA is highly abundant in liver and other tissues involved in lipid metabolism, whereas LXRβ-encoding mRNA is present in all tissues examined (Figure1).
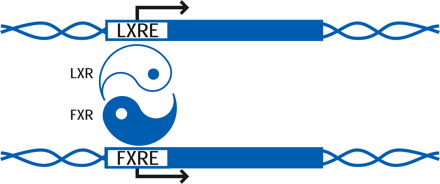
Both LXRα and LXRβ form obligate heterodimers with the retinoid X receptor (RXR) to result in ligand-activated transcription factors that can be activated by ligands (i.e., lipids) for either RXR or LXR (5). RXR–LXR heterodimers bind to a specific DNA sequence called the LXR response element (LXRE), which consists of two hexanucleotide sequences separated by four bases (Figure 1⇓) (6).
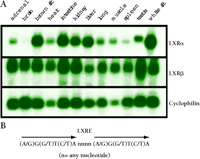
Characterization of oxysterol receptors LXRα and LXRβ.
A. Tissue distribution of LXRα and LXRβ. Shown is a gel blot analysis of adult male mouse tissue mRNA (5μg) probed with DNA fragments that encode sequences of LXRα, LXRβ, or cyclophilin as control.
B. The consensus DNA sequence that defines the LXR response element (LXRE) to which functional LXRs bind to promote transcription. The LXRE consists of a direct repeat of the indicated hexanucleotide sequence separated by four bases.
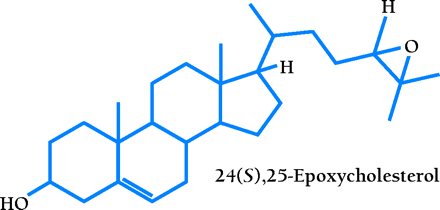
A comprehensive ligand searching strategy identified a specific group of naturally occurring oxysterols as LXR agonists (7,,8). These agonists are intermediates in cholesterol metabolic pathways of the liver, adrenal glands, and brain. The most potent agonists are 24(S),25-epoxycholesterol, which is formed from squalene in a shunt pathway of cholesterol biosynthesis and is abundant in the liver; 22(R)-hydroxycholesterol, which is a transient intermediate in steroid hormone synthesis; and 24(S)-hydroxycholesterol, which is present in the brain (6). A scintillation proximity assay demonstrates that these compounds bind directly to the LXRs with Kd values ranging from 0.1 to 0.4 μM (9,,10). Recently, it was found that 27-hydroxycholesterol is an endogenous ligand for LXRs in cholesterol-loaded human macrophage cells, with Kd values of 85 and 71 nM for LXRα and LXRβ, respectively (11). In addition, nonsteroidal synthetic compounds have been identified as high-affinity LXR-specific agonists (12,,13).
To study the biological function of LXRs, mouse models lacking LXRα, LXRβ, or both LXRs have been generated by targeted gene disruption (14, 15; J. Lobacarro and D. Mangelsdorf, unpublished data). These mouse models have revealed the key role of LXRs in regulating the expression of genes involved in cholesterol catabolism, absorption, and transport, as well as fatty acid synthesis (Table 1⇓). The purpose of this review is to discuss recent and ongoing studies into the regulatory role of LXRs in various aspects of lipid homeostasis.
LXR Regulation of Genes Involved in Lipid Metabolism
CHOLESTEROL HOMEOSTASIS
Bile Acid Synthesis
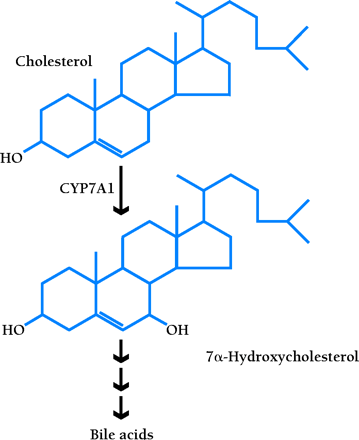
Cholesterol 7α-hydroxylase (CYP7A1) is the rate-limiting enzyme of the classic pathway that converts cholesterol into bile acids (16,,17). The soluble bile acids, primarily produced in the liver, promote the secretion of cholesterol into bile for excretion in feces. In some species (e.g., rodents), expression of the Cyp7a1 gene is induced in response to dietary cholesterol, thereby accelerating the conversion of cholesterol into bile acids so as to promote the net excretion of cholesterol. Lxrα null mice, however, lose the ability to upregulate Cyp7a1 gene expression, and thus accumulate cholesterol esters in the liver, which eventually results in impaired hepatic function (14). Even though LXRβ is present in the liver of these mice, it cannot compensate for the loss of LXRα in regulating CYP7A1 expression (8,,14). Accordingly, Lxrβ null mice maintain normal hepatic cholesterol metabolism and are thus resistant to dietary cholesterol (15; J. Lobacarro and D. Mangelsdorf, unpublished data); however, Lxrα/Lxrβ double-knockout mice have an exacerbated liver phenotype (J. Lobacarro and D. Mangelsdorf, unpublished data).
LXRα upregulates the transcription of Cyp7a1 by directly binding to an LXRE in the promoter of this gene (8,,14). Furthermore, the liver-specific expression of CYP7A1 requires LRH-1 (i.e., liver receptor homolog-1; also called CPF and FTF), a monomeric orphan nuclear receptor (5). The transcription of Cyp7a1 is also regulated via feedback inhibition. Specifically, the bile acid receptor FXR (i.e., farnesoid X receptor) (18,,19) binds bile acids and induces the expression of SHP (i.e., small heterodimer partner), an orphan nuclear receptor that preferentially dimerizes with LRH-1 and represses a number of enterohepatic genes involved in synthesis and transport of bile acids, including Cyp7a1. In this way, LXR and FXR act as the yin and yang of bile acid metabolism, operating together to tightly regulate bile acid homeostasis, respectively functioning, with a cholesterol precursor and cholesterol metabolite, to up- and downregulate Cyp7a1 (Figure 2⇓). Induction of the Cyp7a1 by dietary cholesterol occurs in both the mouse and rat, but not in the human or hamster (4), which suggests that disparate regulatory pathways may exist among species.
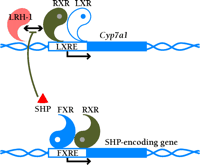
LXR and FXR as the regulatory yin and yang of intracellular cholesterol turnover. The RXR–LXR dimer becomes a functional transcription factor when either protomeric constituent binds ligand; the blue circle associated with the LXR in the schematic represents 24(S),25-epoxycholesterol. The dimer therefore binds to its cognate nucleotide sequence (i.e., the LXRE) in the promoter of Cyp7a1, a gene crucial to cholesterol catabolism. Activation of the LXRE by the dimer is enabled through an interaction with LRH-1. As cholesterol is shuttled into bile acid (represented by white circle) production, FXR becomes functional as a transcription factor in the expression of the SHP-encoding gene. SHP binds to LRH-1 and limits the effectiveness of the RXR–LXR dimer. For a list of genes that are activated by LXR, see Table 1.
Cholesterol Absorption
Dietary and secreted biliary cholesterol enters the intestinal lumen and is absorbed by proximal enterocytes. In the mouse, LXR and RXR agonists, which are metabolic indicators of active cholesterol turnover, decrease cholesterol absorption (12). This net reduction in cholesterol uptake is associated with an RXR/LXR-mediated upregulation of LXRE-containing genes (20–,22) that encode ATP-binding cassette (ABC) transporters. ABC transporters are transmembrane proteins that utilize the energy of ATP hydrolysis to move substrates across cell membranes (23).
In the small intestine, at least three ABC transporters are transcriptionally regulated by LXRs (12,,24) and putatively limit cholesterol absorption by pumping cholesterol back into the lumen of the gut. ABCA1 is a full transporter protein containing two symmetric halves, each composed of a six-transmembrane domain and an ATP-binding cassette. Mutations in the ABCA1 gene result in Tangier disease, which is characterized by a low concentration of plasma high-density lipoprotein (HDL) and the accumulation of cholesterol esters in tonsils, liver, spleen, intestinal mucosa, and macrophage foam cells (25–,27). ABCA1 is upregulated by LXRs in both intestine and macrophages (12). In macrophages, ABCA1 facilitates reverse cholesterol transport (discussed below).
ABCG5 and ABCG8 are half-transporters, each composed of a single transmembrane domain and ATP-binding cassette (24), and are co-expressed exclusively in the liver and intestine. Mutations in either gene cause a rare autosomal recessive disorder called sitosterolemia, which is characterized by increased absorption of cholesterol and toxic plant sterols, and decreased biliary sterol secretion. In vivo and in vitro experiments indicate that ABCG5 and ABCG8 are direct targets of LXRs (24, 59). These findings strongly support the hypothesis that LXRs promote cholesterol loss by increasing biliary cholesterol secretion and limiting cholesterol absorption.
Reverse Cholesterol Transport
The elimination of cholesterol through bile acid production occurs exclusively in the liver, so that excess cholesterol from most other tissues must be returned to the liver, in a process termed “reverse cholesterol transport,” for catabolic elimination. This process is especially important in macrophages, because excess cholesterol in these cells can lead to their conversion into foam cells, thereby promoting atherogenesis. Two ABC transporters, ABCA1 and ABCG1, are upregulated by LXR and RXR agonists in macrophages (12,28,,29). As discussed above, ABCA1 is upregulated by LXRs to result in cholesterol efflux. ABCG1, a half-transporter protein, has also been implicated in the cellular efflux of cholesterol and phospholipid (30). ABCG1, identified by a subtractive hybridization technique as a gene induced during macrophage foam cell formation (29), is also an LXR target gene (28,,29), and contains two functional LXREs (31). A novel ABC half-transporter, ABCG4, has also been characterized (20,32,,33); it shares high sequence identity with ABCG1. Although still controversial, expression of ABCG4 is believed to be upregulated in response to oxysterols and retinoids in human macrophages, suggesting a possible role for this transporter in macrophage lipid homeostasis (33). To date, five members of the ABC transporter superfamily have thus been implicated as downstream mediators of LXR action and play important roles in cholesterol homeostasis.
In addition to dependence upon ABCA1 to make cellular cholesterol available to HDL particles, the primary serum transporters of cholesterol back to the liver, reverse cholesterol transport also relies on LXRs to induce the transcription of apolipoprotein E (apoE), a component of some nascent HDL particles (34). Free cholesterol is thereby esterified within HDL particles and either transported back to the liver for further catabolism and excretion, or transferred to other lipoproteins. In humans, this latter process is mediated by cholesterol ester transfer protein (CETP) (35). Not surprisingly, the gene for CETP has also been reported as a direct target of LXR (36).
LXRs have also been shown to govern the expression of lipoprotein lipase (LPL) (37). LXR facilitates the expression of LPL selectively in liver and macrophages, but not in other tissues that express LPL, so that a previously unsuspected role for LPL is implicated in reverse cholesterol transport. LPL was first characterized as the rate-limiting enzyme in triglyceride hydrolysis in circulating large lipoprotein particles (38). Depending on the system studied, LPL has been reported to have both pro-atherogenic and anti-atherogenic effects. One reason why LXR may upregulate LPL in macrophages would be in order to help clear serum of cholesterol-rich lipoproteins by remodeling triglyceride-rich chylomicrons and very low–density lipoproteins into chylomicron remnants and low-density lipoprotein for uptake by macrophages. In the macrophage, cholesterol esters could then be converted into oxysterols and free cholesterol for efflux and transport back to the liver by HDL. LPL can facilitate HDL-cholesterol uptake into liver cells by a mechanism that does not require the catalytic activity of the lipase (39). LPL expression also promotes maturation of HDL in mice (40).
Throughout the mechanisms of reverse cholesterol transport presented above, LXRs are implicated as key sensors of sterol metabolism, maintaining normal cholesterol balance by promoting sterol efflux from peripheral cells, increasing circulating HDL-cholesterol, and ultimately increasing hepatic sterol catabolism and excretion. LXR regulation of reverse cholesterol transport also suggests a role for LXRs in protecting against atherogenesis, a hypothesis that has been confirmed by studies of Apoe -/- mice in which activation of LXR reduced atherosclerosis (41).
FATTY ACID SYNTHESIS
Depletion of LXRs not only disrupts the metabolism of cholesterol, but also affects fatty acid synthesis (14). The expression of several genes encoding crucial enzymes of this important lipid metabolic pathway is altered in Lxr null mice. Moreover, a role for LXRs in fatty acid metabolism is compellingly indicated from the finding that sterol regulatory element-binding protein-1c (SREBP-1c) is a direct target of LXR action (42–,44). SREBP-1c is a transcription factor that governs the expression of enzymes involved in fatty acid synthesis (45–,47). In Lxr knockout mice, the basal expression level of SREBP-1c is reduced substantially and does not respond to LXR agonists; as a consequence, the transcription of a number of fatty acid synthesizing enzymes fails to be upregulated in response to LXR-specific agonists (14,42–,44). These enzymes include fatty acid synthase (FAS), the central enzyme in de novo lipogenesis, catalyzing all the steps in the conversion of malonyl-CoA to palmitate. A second enzyme that depends on LXR activation via SREBP-1c is stearoyl-CoA desaturase-1 (SCD-1), which catalyzes the Δ9-cis desaturation of stearoyl-CoA and palmitoyl-CoA to oleoyl-CoA and palmitoleoyl-CoA, respectively. Although the upregulation of both enzymes is considered an indirect effect of LXRs, presumably by increasing the transcription and cleavage of SREBP-1c (43), future studies are needed to address whether these and other genes are also directly regulated by LXRs.
Why do the LXRs, which act in many contexts to limit cellular cholesterol content, function to activate the SREBP-1c regulatory network of lipogenesis? One reason may be to coordinate levels of fatty acids and phospholipids, components that are essential to the mobilization of free cholesterol. For example, oleoyl-CoA, a product of SCD-1 catalysis, is the preferred substrate for cholesterol esterification. Therefore, by increasing SCD-1 activity via SREBP-1c, the LXRs would ultimately promote the esterification and storage of free cholesterol within the cell and protect the organism from toxic cholesterol accumulation. In addition, phospholipids facilitate bile acid flow, lipoprotein transport of excess cholesterol, and help maintain the appropriate ratio of cholesterol to other lipids in plasma membranes (42,,44). Thus, the finding that LXRs function to promote lipogenesis provides a conceptual link between sterol and fatty acid metabolic pathways. Unfortunately, pharmacological activation of the LXR/RXR heterodimer can increase lipogenesis to potentially harmful levels, causing hypertriglyceridemia (43). If LXRs are to be considered as therapeutic targets within the context of cholesterol metabolism, their role in triglyceride metabolism must be addressed. The development of novel, gene- and tissue-selective LXR agonists, in a fashion similar to that accomplished in the development of selective estrogen receptor modulators (SERMs), may hold a key to the pharmacological control of cholesterol levels.
HUMAN LXRα AUTOREGULATION
Many nuclear receptors autoregulate their own expression. A classic example of this type of regulation is exhibited by the retinoic acid receptor RARβ. The RARβ2 gene promoter contains an RAR response element that is strongly responsive to retinoic acid (48–,50). Recently, an autoregulatory function has been ascribed to the human LXRα gene (51,,52). This regulation is mediated through an LXRE in the distal portion of the LXRα gene promoter that confers responsiveness to both LXRα and LXRβ. Although this region is conserved in both the mouse and human promoter, autoregulation is only observed in human cells, suggesting that humans may be more responsive to LXRα agonists than mice. Interestingly, one study identified LXRα autoregulation exclusively in macrophages (52), whereas a second study has found autoregulation in adipocytes and hepatocytes (51). The reason for the discrepancy is not clear and calls for further investigation. Nevertheless, the self-induction of LXRα in macrophages in response to lipid loading provides a means for amplifying the effects of oxysterols in reverse cholesterol transport.
LXRα REGULATION BY PPARS
In addition to autoregulatory control, recent studies have shown that the expression of LXRα is also regulated by members of another family of nuclear receptors, the peroxisome proliferator-activated receptors (PPARs). Two subtypes of the PPARs, PPARα and PPARγ, are particularly interesting in that they have been well characterized and are involved in lipid metabolism (53). PPARα is abundantly expressed in liver, kidney, heart, and muscle, and regulates genes involved in fatty acid catabolism and lipoprotein production. PPARγ is selectively expressed in adipose tissues and governs adipogenesis. Both PPAR subtypes are found in macrophages that are associated with atherosclerotic plaques.
PPARs promote cholesterol efflux in macrophages through an ABCA1 pathway, possibly by mediating expression of LXRα (54,,55). A PPAR response element (PPRE) has been identified in both the mouse and human LXRα gene (51,,55). Although one study claims that both PPARα and PPARγ agonists can induce LXRα expression in macrophages (54), a second study shows that only PPARγ can upregulate expression of LXRα through the PPRE (55). It has also been reported that PPARα agonists induce the expression of LXRα in rat liver (56); however, this observation could not be corroborated (55). Interestingly, no regulation of LXRβ has been observed thus far in any studies, implicating distinct roles of LXRα and LXRβ in certain regulatory pathways. Nevertheless, the transcriptional regulation of LXRα by PPARs suggests an overlap between LXR and PPAR signaling pathways. This regulatory relationship, especially in regard to macrophages, may have important therapeutic implications for the treatment of atherosclerosis.
OTHER POTENTIAL ROLES OF LXRs
Subtractive hybridization between wild-type and Lxrα null mice has revealed the existence of several somewhat surprising targets of LXR transactivation. One such targeted gene (denoted as YZ-2 in Table 1⇑) encodes a novel, liver-specific protein that shares identity with uridine phosphorylase (Y. Zhang and D. Mangelsdorf, unpublished observation), which is one of the two enzymes involved in the pyrimidine salvage pathway and catalyzes the reversible phosphorolysis of uridine to produce uracil (57). Interestingly, the expression pattern of this novel gene is strikingly similar to that of another LXR-targeted gene, CYP7A1. The novel analog of uridine phosphorylase is expressed, according to a distinct circadian rhythm that also typifies expression of CYP7A1, primarily in the liver and is induced in response to LXR agonists; its expression is reduced in Lxr null animals. Although direct regulation by LXR has not yet been established, this gene is transcriptionally regulated by the orphan nuclear receptor HNF4 (i.e., hepatocyte nuclear factor 4). The metabolism of uridine nucleotides is involved in a variety of biological functions, including cell growth, and sugar and lipid metabolism (58). The regulation of this gene by LXR raises the possibility that LXRs are involved in pyrimidine-dependent metabolic pathways.
As shown in Figure 1⇑, LXRs are also expressed in many tissues other than liver. One site of LXR production that is particular intriguing is the adrenal gland, where a rich supply of cholesterol serves as the precursor to all steroid hormones. Interestingly, several intermediates in the steroidogenic pathway (e.g., 20(S)-hydroxycholesterol and 22(R)-hydroxycholesterol) are potent LXR activators. The function of LXRs in the adrenal gland is not known, but they may serve some role as sterol sensors in regulating availability of cholesterol for steroid synthesis. In some tissues, LXRβ is more abundant than LXRα, raising the possibility of disparate, tissue-selective roles for the two LXR types. For example, brain predominantly expresses LXRβ and synthesizes the potent LXR activator 24(S)-hydroxycholesterol. Although no phenotypic consequence has been specifically observed with respect to the brain in Lxr knockout animals, the high abundance and requirement of cholesterol in brain may well be supported by mechanisms involving LXRs.
CONCLUSIONS AND THERAPEUTIC IMPLICATIONS
The use of Lxr null mice and LXR-selective agonists has identified crucial genes regulated by LXRs, and establishes the important role of LXRs as sterol sensors that govern the absorption, transport, and catabolism of cholesterol (Figure 3⇓). These studies suggest LXRs as exciting therapeutic targets for novel treatment of hypercholesterolemia and atherosclerosis. However, one potential drawback of attempts to exploit LXR agonism is hypertriglyceridemia caused by upregulation of SREBP-1c. Therefore, one goal of future studies should be focused on separating these two effects, possibly by designing LXRα- and LXRβ-specific agonists. Several lines of evidence suggest that LXRα and LXRβ have some redundant, but also many distinct functions. For example, in vivo studies show that upregulation of LPL by synthetic LXR ligands is more sensitive to the presence of LXRα than LXRβ (37). Newer studies also support the concept that the LXRα gene is transcriptionally regulated in several tissues (36,51,52,55,,56). In contrast, very little is known about the precise role of LXRβ, which is expressed abundantly in a myriad of tissues (Figure 1⇑). Clearly, one area of future study will be in differentiating the roles of LXRα and LXRβ . The design of LXRα- and LXRβ-selective agonists and identification of LXR subtype–specific pathways may aid in the development of novel therapeutic approaches to fight cholesterol and lipid disorders.
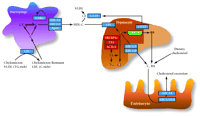
Model describing the LXR regulatory pathways governing lipid metabolism.
In liver, intestine, and macrophages, LXRs regulate the expression of genes involved in bile acid synthesis (green box), fatty acid synthesis (red boxes), and cholesterol efflux and transport (blue boxes). LXRs sense intracellular cholesterol levels to help maintain cholesterol homeostasis by increasing bile acid excretion, reducing cholesterol absorption, and increasing cholesterol transport back to the liver for catabolism and storage. (C, cholesterol; CE, cholesterol ester; BA, bile acid; FA, fatty acid; TG, triglyceride; HDL, high density lipoprotein; VLDL, very low density lipoprotein; LDL, low density lipoprotein. See text for details.)
Acknowledgments
We thank Dr. Joyce J. Repa for her advice and suggestions during the preparation of this manuscript. We thank the Mango lab for their criticism and support. YZ is a postdoctoral fellow in the Department of Pharmacology at the University of Texas Southwestern Medical Center. DJM is an investigator of the Howard Hughes Medical Institute. This work was also supported by grants from the Robert A. Welch Foundation, the NIH, and the Human Frontier Science Program.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

David Mangelsdorf, PhD, is Professor of Pharmacology and Adjunct Professor of Biochemistry at the University of Texas Southwestern Medical Center at Dallas, and an Associate Investigator of the Howard Hughes Medical Institute. He received his undergraduate degree from Northern Arizona University in Flagstaff before pursuing doctoral work in biochemistry at the University of Arizona in Tucson. Before joining UT Southwestern in 1993, he worked as a postdoc on the retinoid X receptor at the Salk Institute. Dr. Mangelsdorf has received several honors including the ASPET John J. Abel Award, the Endocrine Society Richard A. Weitzman Memorial Award, and the Falk Foundation's Adolf Windaus Prize.
Yuan Zhang, PhD, received her undergraduate training at the University of Science and Technology of China in Hefei. In 2001, Dr. Zhang completed her doctoral work in the Mangelsdorf laboratory at UT Southwestern, where she is now continuing her research as a postdoctoral fellow.

