Addiction: Making the Connection Between Behavioral Changes and Neuronal Plasticity in Specific Pathways
Abstract
There is an emerging consensus that drug addiction is a form of maladaptive learning. Drugs of abuse usurp the neuronal circuitry involved in motivation and reward, leading to aberrant engagement of learning processes. As a result, drug-associated cues can trigger craving and compulsive drug-seeking behavior, and voluntary control over drug use is lost. Abused drugs can also modulate long-term potentiation (LTP) and long-term depression (LTD) in neuronal circuits associated with the addiction process, suggesting a way for the behavioral consequences of drug-taking to become reinforced by learning mechanisms. This review will assess progress in correlating these effects on LTP and LTD with behavioral changes in animal models of addiction, particularly behavioral sensitization.
The image evokes the alluring yet sinister nature of cocaine. By usurping neuronal pathways normally involved in motivated
behavior, cocaine promotes “learning” of compulsive behavioral responses that underlie drug craving and addiction.
Introduction
Cocaine and amphetamines enter the brain and interfere with the function of cell membrane transporters that normally remove dopamine and other monoamines (serotonin and norepinephrine) from the synapse after these transmitters are released. This interference produces a rapid elevation of dopamine levels that results in psychomotor stimulation: a feeling of being “high.” Although these interactions are becoming better understood, it remains a mystery how this initial elevation in dopamine levels leads, in some people, to a compulsive pattern of drug-seeking and drug-taking behavior. Even if abstinence is achieved, cocaine addicts remain vulnerable for years to episodes of craving and relapse triggered by stimuli previously associated with drugs (1,,2). This persistent vulnerability to drug-conditioned stimuli is also observed in animal models of addiction (3).
These features of addiction suggest that it may be an exceptionally powerful form of neuronal plasticity, which can be broadly defined as the ability of the nervous system to modify its response to a stimulus based on prior experience, and is believed to underlie learning and memory. Plasticity might also underlie addiction, because signaling through glutamate, the key neurotransmitter for producing and maintaining synaptic plasticity, is important for the formation of behavioral sensitization—a prominent animal model of addiction (4,,5). This role for glutamate receives further support from imaging studies of the brains of human cocaine addicts: stimuli previously associated with drug use (e.g., drug paraphernalia) trigger intense drug craving, and at the same time, activate glutamate-rich neuronal circuits implicated in learning and memory (6). Animal studies implicate the same glutamate-rich circuits, and suggest that the progression to addiction is a form of habit-based learning (7). Parallels between addiction and learning are reflected in the striking similarities between the signal transduction cascades and molecular adaptations associated with both processes (8). For example, chronic administration of amphetamine or cocaine produces dramatic changes in the morphology of dendritic spines in addiction-related brain regions, which closely resemble those associated with learning (9,,10).
Addiction and Long-Term Potentiation
Learning and memory are thought to be encoded by changes in the usage of interneuronal connections (11). In other words, synapses that experience frequent use will be strengthened, whereas those that receive less use are weakened. In this way, an experience (which results in activation of pathways and increased synaptic “use”) may produce a long-lasting enhancement of communication between neurons in those pathways (resulting in a memory). For thirty years, long-term potentiation (LTP) has been the most compelling model for studying the synaptic basis of use-dependent changes in the strength of interneuronal connections. In LTP, brief repetitive activation of excitatory glutamate-containing pathways (produced by high frequency stimulation, or tetanus) leads to an increase in the strength of glutamate synapses that lasts many hours in brain slices and even for weeks in intact animals (12,,13). Thus, a test stimulus applied to a pathway before the tetanus will produce a postsynaptic response of a certain magnitude, whereas the same test stimulus applied to a pathway after tetanus will produce a larger response. Subsequent studies showed that weaker patterns of stimulation produce an opposite change, termed long-term depression (LTD).
The cellular basis of LTP and LTD continues to be the focus of intense investigation. In general, an N-methyl-d-aspartate (NMDA) receptor–mediated increase in postsynaptic calcium concentrations is required for the development of both LTD and LTP, with small increases in calcium preferentially activating protein phosphatases and producing LTD, whereas larger increases in calcium preferentially activate protein kinases and produce LTP (14). The expression of LTP involves enhanced transmission through the α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor in the postsynaptic cell. Important mechanisms contributing to the formation of LTP include phosphorylation of the AMPA receptor and increased insertion of AMPA receptors at synaptic membranes, whereas AMPA receptor dephosphorylation and internalization may contribute to LTD (15,16,,17).
LTP was first demonstrated in the hippocampus, a brain region important for learning, which was part of the reason that LTP was speculated to underlie memory formation. However, many investigators now question whether LTP plays a simple role in learning and memory—particularly the kinds of complex associative learning that contribute to drug-seeking behavior and relapse (18). Alternatively, LTP (and LTD) occur in many brain regions other than the hippocampus, suggesting that these phenomena represent general mechanisms for altering synaptic strength in response to changes in synaptic use (regardless of whether this directly underlies memory formation). We have hypothesized that drugs of abuse alter LTP or LTD in neuronal pathways, by altering activity in networks related to motivation and reward, or influence the ability of “normal” patterns of activity to produce appropriate forms of LTP and LTD (19). Abnormal engagement of LTP or LTD may be the first step in the cascade leading to structural changes that underlie persistent modifications in brain function (20). Recent studies have provided general support for this hypothesis, showing that drugs of abuse can, in fact, modify or induce LTP and LTD in neuronal pathways related to addiction.
This review focuses on the challenging task of making connections between behavioral changes related to cocaine and amphetamine addiction and LTP or LTD in specific neuronal circuits. After reviewing the neuronal circuits involved in drug action, we then consider how addiction may be related to changes in neuronal pathways converging in the nucleus accumbens. We know that these pathways are important for addiction, but we are just beginning to understand how drugs of abuse modify their activity. Additionally, we will consider plasticity in the ventral tegmental area (VTA). Here, we are closer to making connections between plasticity and specific changes in neuronal activity that contribute to an animal model for addiction, behavioral sensitization.
Neuronal Pathways Important In Addiction
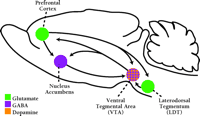
The nucleus accumbens (also termed ventral striatum) plays a central role in the neuronal circuits that are responsible for motivated, goal-directed behaviors—in other words, the type of behaviors that underlie compulsive drug-seeking in cocaine addicts (Figure 1⇓). Goal-directed behaviors are produced by glutamate-containing projections that originate in limbic and cortical regions [most importantly, the basolateral amygdala (BLA), the hippocampus and the prefrontal cortex] and converge on a common postsynaptic target: the medium spiny neuron of the nucleus accumbens. The output of these nucleus accumbens neurons, conveyed through their projections to the ventral pallidum and ventral mesencephalon, is responsible for motor execution of these goal-directed behaviors. Thus, the nucleus accumbens serves as an interface between limbic and motor systems (21,,22), and ultimately, drug-seeking behavior depends on glutamate transmission in the nucleus accumbens (23,,24).
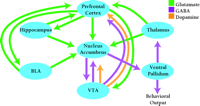
The neural circuitry involved in drug addiction. The nucleus accumbens, an interface between limbic and motor systems, plays a central role in this circuitry. Goal-directed behaviors are elicited by glutamate projections from limbic and cortical regions to the nucleus accumbens, while projections from the nucleus accumbens to motor regions are responsible for motor execution of these behaviors. Dopamine neurons located in the VTA innervate many limbic and cortical regions important for addiction, including nucleus accumbens and prefrontal cortex. Dopamine and glutamate inputs typically converge on common postsynaptic targets. BLA, basolateral amygdala; VTA, ventral tegmental area. [Based on (7).]
Dopamine has long been the focus of addiction research because cocaine and other stimulants act initially by increasing the release of dopamine in the brain, and because dopamine neurons participate in the elicitation of normal behaviors related to motivation and reward. Indeed, dopamine is readily integrated into glutamate-based models for addiction because glutamate- and dopamine-containing pathways converge in many brain regions, including the nucleus accumbens (Figure 1⇑). Thus, the same nucleus accumbens neurons that receive limbic and cortical glutamate inputs also receive dopamine inputs from the VTA (25). Dopamine regulates interactions between glutamate inputs to nucleus accumbens neurons (26,,27), both by directly influencing synaptic transmission and by modulating voltage-dependent conductance (28), in a manner complicated by the ability of the same glutamate inputs to influence dopamine transmission (29). VTA dopamine neurons also project to limbic and cortical regions important for addiction, such as the prefrontal cortex and the amygdala. Dopamine transmission in these regions modulates the activity of addiction-related circuits (30) and is necessary for some types of drug-seeking behavior (31,32,,33).
Nucleus Accumbens Glutamate and Drug-seeking Behavior
Animal models have been used extensively to study the circuits and neurotransmitters involved in drug-seeking behavior. Rats learn to associate drug availability with a stimulus (such as a flashing light or a particular environment), and will perform conditioned behaviors that are related to drug seeking when presented subsequently with that stimulus. Rats can also learn to self-administer drugs (e.g., by pressing a bar that delivers an intravenous drug infusion) in response to a priming injection of drug or a conditioned stimulus that was previously associated with drug availability.
Different glutamate inputs to nucleus accumbens neurons have different functions in drug-seeking behavior. Projections from the BLA to the nucleus accumbens are important for learned associations between stimuli and drug reward, and for the expression of motor behavior that is driven by motivationally relevant stimuli. These projections play a key role in drug-seeking behavior elicited by discrete cues that were previously paired with drug exposure (34,,35). On the other hand, drug-seeking behavior triggered by injection of a low priming dose of cocaine does not require the amygdala—perhaps because this involves a response to drug itself, rather than to a conditioned stimulus—but does require a circuit that includes the VTA, prefrontal cortex, nucleus accumbens, and ventral pallidum (31,,34). The hippocampus may be involved in conditioning to general contexts, such as a particular cage or room environment previously associated with drug availability. The direct and indirect connections of the hippocampus to the nucleus accumbens may enable such contextual cues to influence drug-seeking behavior (36), although the role of the hippocampus in cue-elicited craving has been studied far less extensively than that of the amygdala. The prefrontal cortex is implicated in executive control of behavioral output based on stimulus value and expected outcome, and plays an important role in animal models of addiction (37). Excitatory inputs from the prefrontal cortex, BLA, and hippocampus are integrated by dopamine to determine the net level of excitatory output to neurons in the nucleus accumbens (26,,38).
What Underlies The Transition to Addiction?
The dopamine neurons that participate in motivation and reward have their cell bodies in the VTA and project to a number of limbic and cortical brain regions, including the nucleus accumbens. These dopamine neurons are activated by natural, rewarding stimuli such as food or sex, resulting in increased dopamine release in target brain regions. Activation of dopamine-releasing neurons is important in predicting the likelihood of reward when an animal is presented with reward-related stimuli (39). Thus, one normal function of dopamine may be to help consolidate stimulus-response learning, so individuals acquire the habit of pursuing those stimuli that are both rewarding and necessary for survival.
If the same neuronal circuits are involved in responding for “good rewards” such as food or sex, and “bad rewards” such as cocaine, why is it that cocaine, but usually not food or sex, leads to addiction? The answer is probably related to the fact that cocaine and amphetamines increase dopamine release in a more prolonged and unregulated manner than natural stimuli, because these drugs interact with the dopamine transporter and interfere with the removal of dopamine from the synaptic cleft. Over the span of several repeated drug exposures, compensatory changes occur in dopamine-receptive neurons that presumably start a chain reaction of events that ultimately influences basic mechanisms of synaptic plasticity. Possible mechanisms involved in this process have been reviewed recently (19). The important point is that drugs of abuse usurp normal learning mechanisms and thereby “cement” behavioral responses related to drug-seeking behavior. In some people, this progresses to a form of habit-based learning so strong that it persists even in the face of tremendous adverse personal consequences (7).
The behavioral changes that underlie the transition from drug experimentation to compulsive drug-seeking behavior may be due to synaptic plasticity in the neuronal circuits depicted in Figure 1⇑. Based on what is known about the role of particular pathways in drug-seeking behavior, we can speculate about how plasticity in particular pathways might contribute to various aspects of addiction (Figure 2⇓). For example, strengthening the synaptic connections between the amygdala and the nucleus accumbens may underlie the abnormally strong stimulus-reward learning that occurs in addiction, enabling drug-associated cues to elicit craving and relapse even after long periods of abstinence. This increase in conditioned control of behavior may be enhanced by sensitization of brain systems to the incentive-motivational effects of drugs, an effect that may underlie the transition from “wanting” to “craving”, and at least initially, may involve LTP in VTA dopamine neurons. Another important change may involve an increase in impulsive behavior owing to the loss of inhibitory control mechanisms. The prefrontal cortex is responsible for cognitive functions related to working memory and planning. Normally, descending projections from the prefrontal cortex (to the nucleus accumbens, amygdala and other brain regions) exert inhibitory control over reward-seeking behaviors. Recent evidence, from both rat and human studies, indicates that chronic cocaine exposure impairs these inhibitory control mechanisms. Decreased inhibitory control, combined with enhanced responsiveness to drug-related cues and sensitization of “drug wanting”, may explain the compulsive nature of drug-seeking behavior in addicts (7,35,,37).
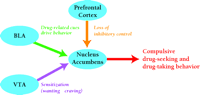
Possible relationships between neuronal plasticity in particular pathways and behavioral changes that lead to addiction. Several types of behavioral changes may contribute to compulsive drug-seeking and drug-taking behavior in addicts. Alterations in transmission between the BLA and the nucleus accumbens may strengthen stimulus-reward learning, increasing the ability of drug-related cues to control behavior. Sensitization of the incentive-motivational effects of drugs, due to alterations in transmission between the VTA and the nucleus accumbens, may transform drug “wanting” to “craving”. Alterations in transmission between the prefrontal cortex and limbic targets, including nucleus accumbens, may lead to impairment of inhibitory control mechanisms that normally govern reward-seeking behavior. Ultimately, drug-seeking behavior depends on glutamate transmission in the nucleus accumbens. BLA, basolateral amygdala; VTA, ventral tegmental area. [Based on (37).]
Does intensification of stimulus-reward associations reflect LTP in pathways between BLA and nucleus accumbens? Does loss of inhibitory control reflect LTD in pathways between prefrontal cortex and its limbic targets? While it is not yet possible to answer these questions, recent studies have made significant progress towards characterizing LTP and LTD in reward-related brain regions, including the prefrontal cortex (40,,41) and amygdala (42). The remainder of this article will consider the role in addiction of synaptic plasticity in the nucleus accumbens and the VTA.
Synaptic Plasticity in the Nucleus Accumbens
Both LTP and LTD are produced in the nucleus accumbens in response to repetitive stimulation of projections from the prefrontal cortex (43). LTP is also produced by repetitive stimulation of hippocampal projections to the nucleus accumbens (44). NMDA receptor activation is required for both LTP and LTD in nucleus accumbens neurons (43,45,,46). While LTP may be modulated by dopamine, LTD is not affected by application of dopamine (46). It is notable that the properties of LTP and LTD in the nucleus acumbens (ventral striatum) differ from those in dorsal striatum (8,,19), where these processes have been better characterized (47).
Neurons in the nucleus accumbens are normally quiescent, and their activation requires synchronous stimulation of multiple excitatory inputs (38). Therefore, any drug-induced change that either enhances or depresses their response to a particular input will have a very significant influence on their activity. Recent studies suggest that two kinds of changes may occur. First, modulatory effects of stimulants (or dopamine itself) on LTP may be altered after repeated drug administration. Li and Kauer reported that the application of amphetamine blocked the induction of LTP in nucleus accumbens slices prepared from control rats, whereas amphetamine did not block LTP in slices prepared from rats pretreated with amphetamine for six days (48). A relative shift towards LTP that may be attributable to loss of D2 receptor-mediated inhibitory effects has also been reported in striatal slices prepared from rats chronically treated with methamphetamine or ethanol (49,,50). Thus, the response of nucleus accumbens or striatal neurons to drug challenge might be different after repeated drug exposure because inhibitory mechanisms are dampened. The second kind of change may involve an alteration in the strength of particular synaptic connections that long outlasts the period of drug administration. For example, nucleus accumbens slices prepared from mice two weeks after discontinuing repeated cocaine treatment exhibited LTD at excitatory synapses between cortical afferents and nucleus accumbens neurons (51). This observation is consistent with previous work indicating that the nucleus accumbens is less excitable after chronic stimulant exposure (19), and suggests that decreases in the cortical regulation of nucleus accumbens neuronal activity could be related to the loss of inhibitory control that is implicated in addiction and thought to involve prefrontal cortex dysfunction (37). Loss of inhibitory control at the level of the prefrontal cortex may also contribute to the expression of behavioral sensitization (7).
These are exciting findings; nonetheless, we are a long way from understanding the relationship between LTP or LTD in a particular pathway and a specific behavioral change in animal models or human studies of addictive behavior. Part of the difficulty is that we do not yet understand how dopamine and glutamate work together to determine the output of nucleus accumbens neurons under normal conditions (28,,52), let alone after chronic drug administration. In contrast, we are closer to understanding how plasticity in the VTA may contribute to behavioral sensitization, an animal model of addiction.
Glutamate and Behavioral Sensitization
Behavioral sensitization refers to the progressive enhancement of species-specific behavioral responses that occurs during repeated drug administration and persists even after long periods of withdrawal. Sensitization occurs in humans (53), but most studies have focused on sensitization of locomotor activity in rodents. There is compelling evidence that locomotor sensitization provides a useful model for sensitization of drug craving. Both depend on VTA dopamine neurons projecting to limbic and cortical regions (Figure 1⇑). Prior exposure to cocaine or amphetamine, resulting in locomotor sensitization, promotes drug self-administration and enhances stimulus-reward learning and responding for conditioned reward (7). Finally, both sensitization in rats and drug craving in humans are strongly modulated by environmental stimuli, conditioning and stress (54,,55). Once established, sensitization is very long-lasting—amphetamine sensitization can last up to a year in rats, a species that lives only one or two years (56)—and is accompanied by dramatic changes in addiction-related neuronal circuits at molecular, cellular, and systems levels (7,57–,59).
Robinson and Berridge have developed an incentive-sensitization theory of addiction (60,,61) that remains one of the most influential in the field. A key element of this theory is that “liking” and “wanting” are produced by distinct neuronal mechanisms, and it is “wanting” that intensifies with repeated drug use. In other words, repeated drug exposure does not increase the pleasure produced by a drug, but does increase craving. According to this theory, addictive drugs produce long-lasting adaptations in brain systems mediating incentive-motivational effects, leading to sensitization of these systems to drugs and, importantly, to cues that trigger pursuit of rewarding stimuli (62). Sensitization of incentive-motivational effects would promote compulsive behavior, perhaps by contributing to the enhanced stimulus-reward learning discussed above in relation to projections from the amygdala to the nucleus accumbens (Figure 2⇑). In fact, although we will focus on VTA below, the amygdala might also participate in the induction of sensitization (57).
Synaptic Plasticity in the VTA
The circuitry involved in sensitization is complex, because sensitization represents a cascade of events involving different brain regions and different transmitter systems at different withdrawal times. However, microinjection studies have established that the initiation of sensitization to cocaine or amphetamine occurs in the VTA. Thus, direct injection of amphetamine into the VTA has no acute effect on locomotor activity, but repeated intra-VTA administration results in a sensitized locomotor response to systemic injections of amphetamine, cocaine, or morphine. In contrast, sensitization is expressed in the nucleus accumbens, because direct injection of amphetamine into nucleus accumbens is sufficient to elicit a sensitized response in rats that received repeated systemic or intra-VTA injections (57,,58).
The anatomical separation of sites for initiation and for expression implies a “transfer” of sensitization from the VTA to the nucleus accumbens, presumably as a result of changes in the firing rate or pattern of dopamine neurons projecting from VTA to nucleus accumbens. Indeed, the firing rate of VTA dopamine neurons is increased for several days after discontinuing repeated administration of cocaine or amphetamine. Firing rates then normalize within a week or so. This transient activation of dopamine-releasing neurons is believed to be a key step in the development of long-lasting behavioral sensitization. The mechanism underlying the activation may involve increased glutamate transmission in the VTA during drug administration and the early withdrawal period. This idea is consistent with studies showing that the development of sensitization is prevented by blocking glutamate transmission in the VTA, or by lesioning the prefrontal cortex, which sends glutamate projections to the VTA (57,,58).
One way to account for these findings is to propose that repeated drug administration results in LTP at synapses between glutamate nerve terminals and VTA dopamine neurons, leading to a transient increase in the firing rate of dopamine neurons (57). This, in turn, would increase dopamine release in the nucleus accumbens and other addiction-related brain regions that are the targets of dopamine projections (including prefrontal cortex, amygdala and hippocampus). Drug-induced changes in dopamine release may be one of the triggers for the persistent electrophysiological and neurochemical changes observed in these regions after discontinuing drug use (57,,58).
Once it has developed, LTP is expressed as an enhancement of postsynaptic AMPA receptor transmission. Thus, if the initiation of behavioral sensitization is associated with LTP at glutamate synapses on VTA dopamine neurons, and if LTP is responsible for transient activation of dopamine cell firing during early withdrawal, then dopamine neurons in sensitized rats should exhibit increased responsiveness to AMPA at early withdrawal times, but not at later times. This prediction has been confirmed in two ways. First, VTA dopamine neurons recorded from chronic cocaine- or amphetamine-pretreated rats are more responsive to the excitatory effects of AMPA (63). Second, intra-VTA administration of AMPA produces a greater increase in nucleus accumbens dopamine levels in rats previously treated with amphetamine (64). The latter study suggests that the enhancement of AMPA receptor transmission occurs in those VTA dopamine neurons that project to the nucleus accumbens. In both studies, no change in responsiveness to NMDA was observed and the increased responsiveness to AMPA was evident in rats tested three days after discontinuing drug administration but not at later times when dopamine cell firing rates have normalized.
But can a drug regimen that produces sensitization in a whole animal influence synaptic plasticity in the VTA? In prepared midbrain slices from control mice and from mice injected the day before with a single injection of saline or cocaine, the cocaine treatment—which produces “one-shot” behavioral sensitization—produced an enhancement of AMPA receptor transmission in VTA dopamine neurons that was attributable to LTP (65). Cocaine-induced LTP was observed in slices prepared five days, but not ten days, after cocaine injection. Thus, cocaine-induced LTP is a transient phenomenon, similar to the enhancement of AMPA receptor transmission detected in vivo (63,,64). Furthermore, LTP was blocked by an NMDA receptor antagonist, as was sensitization. These findings argue strongly for linkage between cocaine-induced LTP and sensitization.
How do dopamine-releasing psychomotor stimulants promote LTP in the VTA? Amphetamine or dopamine, acting through D2 dopamine receptors, can block the induction of LTD in midbrain dopamine neurons, through a mechanism that may involve inhibition of high threshold calcium channels (46,,66). Inhibiting LTD through cocaine-dependent stimulation of dopamine receptors may increase neuronal excitability and thus promote LTP. LTP may also be promoted by stimulant-induced increases in glutamate levels in the VTA, although exactly how cocaine and amphetamine modulate VTA glutamate levels is a matter of some debate (19). Dopamine receptor stimulation may also increase the excitability of midbrain dopamine neurons by reducing inhibitory effects of metabotropic glutamate receptors (67).
Based on results summarized above, a model for the initiation of sensitization in the VTA should include: 1) a role for LTP induction in VTA dopamine neurons, 2) a key role for VTA dopamine neurons projecting to the nucleus accumbens, and 3) a necessary role for prefrontal cortex (recall that lesions of prefrontal cortex prevent the development of sensitization). The simplest solution would be to propose that the LTP leading to sensitization occurs at synapses between prefrontal cortex terminals and VTA dopamine neurons projecting to nucleus accumbens. Unfortunately, this hypothesis is not consistent with recent anatomical findings. Sesack and colleagues have shown that prefrontal cortex neurons do not make excitatory synapses on dopamine-releasing VTA neurons that project to the nucleus accumbens; however, they do synapse on γ-aminobutyric acid (GABA)-releasing VTA neurons that project to the nucleus accumbens. Conversely, prefrontal cortex neurons synapse on VTA dopamine neurons, but not on VTA GABA neurons, that project back to the prefrontal cortex (68) (Figure 3⇓). Neurochemical findings are consistent with this anatomical arrangement (69).
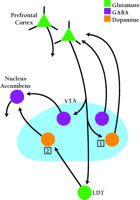
Possible sites for LTP in the ventral tegmental area during the induction of behavioral sensitization. The VTA contains dopamine neurons that project to nucleus accumbens (mesoaccumbens dopamine neurons) and dopamine neurons that project to prefrontal cortex (mesoprefrontal dopamine neurons). Likewise, the VTA contains mesoaccumbens GABA neurons and mesoprefrontal GABA neurons. VTA dopamine and GABA neurons also innervate other brain regions not shown in the diagram. Glutamate projections from the prefrontal cortex synapse on mesoaccumbens GABA neurons but not mesoaccumbens DA neurons, and on mesoprefrontal DA neurons but not mesoprefrontal GABA neurons (68). Some evidence suggests that prefrontal cortex also communicates with mesoaccumbens dopamine neurons indirectly, via projections to the LDT. The LDT sends both glutamate and cholinergic projections to mesoaccumbens dopamine neurons (71). The cholinergic fibers are not shown in the diagram. Based on this anatomy and other evidence, two possible sites are proposed for cocaine-induced LTP leading to the initiation of behavioral sensitization. Site 1: Excitatory synapses between prefrontal cortical projections and mesoprefrontal dopamine neurons. Site 2: Excitatory synapses between LDT projections and mesoaccumbens dopamine neurons. VTA, ventral tegmental area; LDT, laterodorsal tegmentum. [Based on (68).]
An alternative hypothesis is that the LTP important for sensitization occurs at synapses between prefrontal cortex neurons and VTA dopamine neurons projecting to the prefrontal cortex (site 1 in Figure 3⇑), consistent with evidence for a direct excitatory link between these populations. In this model, changes in prefrontal cortex occur first, and then trigger longer-lived changes in the nucleus accumbens through the many interconnections that exist between the two regions (Figure 1⇑). Indeed, the prefrontal cortex is necessary for cocaine-induced adaptations occurring in the nucleus accumbens (70), and there is evidence for changes in both glutamate and dopamine transmission in the prefrontal cortex shortly after discontinuing repeated cocaine or amphetamine administration (7). However, this proposed site for LTP (site 1 in Figure 3⇑) is not consistent with evidence that AMPA receptor transmission is enhanced in those VTA dopamine neurons that project to nucleus accumbens (64).
Another possibility is that VTA dopamine neurons projecting to nucleus accumbens are excited by glutamate (and cholinergic) projections from the laterodorsal tegmentum (LDT) and neighboring mesopontine nuclei (71). Thus, the LTP important for initiating sensitization might occur at synapses between the LDT and VTA dopamine neurons projecting to nucleus accumbens (site 2 in Figure 3⇑). Although promising, this hypothesis raises questions about how the prefrontal cortex is brought into the circuit. The prefrontal cortex projects to the LDT, and it has been proposed that this indirect route to the VTA enables the prefrontal cortex to exert excitatory effects on midbrain dopamine neurons (72). This hypothesis is consistent with experiments showing that the prefrontal cortex activates VTA dopamine neurons through polysynaptic pathways rather than through a direct monosynaptic projection (73) (Figure 4⇓). However, even this indirect route is not consistent with all findings, as a recent study showed that stimulation of the prefrontal cortex at physiologically relevant frequencies actually inhibits dopamine release in the nucleus accumbens (74). Of course, the prefrontal cortex could be brought into the circuit later, after changes have occurred in the pathway from LDT to VTA to nucleus accumbens, through interconnections between these regions (Figure 1⇑).
Addiction-related pathways in the rat brain. The VTA contains both dopamine and GABA neurons that innervate the nucleus accumbens, prefrontal cortex, and other forebrain targets not shown in the diagram. The prefrontal cortex sends glutamate projections to VTA, LDT, nucleus accumbens, and other limbic regions not shown in the diagram (see Figure 1). In the VTA, glutamate inputs from prefrontal cortex synapse on mesoaccumbens GABA neurons and mesoprefrontal DA neurons. Mesoaccumbens dopamine neurons also receive glutamate and cholinergic inputs from the LDT (cholinergic inputs not shown). See legend to Figure 3 for more details.
A cornerstone of the above hypotheses is that the induction of sensitization is prevented by blocking NMDA receptors in the VTA, which also prevents the induction of LTP by systemic cocaine (65), but it is possible that the two are not linked. An alternative possibility is that the important effect of NMDA receptor blockade in the VTA is to alter the activity of VTA GABA neurons that project locally as well as to the nucleus accumbens and other forebrain regions (57,,75). Unlike dopamine neurons, GABA neurons of the VTA do not exhibit LTP under normal conditions (76) or after cocaine treatment (65). While VTA GABA neurons can exhibit LTD (66), effects of cocaine on this process have yet to be examined. Nevertheless, there is evidence that the activity of these GABA neurons is altered by repeated drug administration. For example, GABAB receptor transmission in the VTA is enhanced after withdrawal from amphetamine and other drugs (77). Another exciting finding is that brief (two or three minutes) co-activation of group I metabotropic and NMDA glutamate receptors in the ventral midbrain produced a long-lasting (greater than one hour) enhancement in the rhythmicity of GABAergic inhibitory synaptic activity (78). This effect involved direct electrical coupling of neurons through gap junctions (78), a phenomenon that is altered by chronic amphetamine treatment in other brain regions (79). A role for this mechanism in sensitization would account for the fact that the development of sensitization is inhibited by administration of either NMDA or metabotropic glutamate receptor antagonists into the VTA (80). Alterations in inhibitory tone, which controls basal firing characteristics of VTA dopamine neurons, would be expected to have major effects on the response of these neurons to excitatory inputs (29).
Future Directions
The demonstration that drugs of abuse alter LTP and LTD in reward-related neuronal pathways is a very exciting development. The next challenge is to study LTP and LTD in the context of animal models of drug seeking and relapse. Another challenge is to understand how drugs of abuse influence the cellular mechanisms governing synaptic plasticity. Two mechanisms that contribute to LTP are phosphorylation of AMPA receptors, which increases their activity, and insertion of new AMPA receptors into synaptic sites (15,,17). Signaling through the D1 dopamine receptor may influence both processes. For example, in neurons of the nucleus accumbens, acute stimulation of D1 dopamine receptors increases the phosphorylation of AMPA receptors (81), and there are compensatory changes in AMPA receptor phosphorylation in striatal neurons after chronic cocaine administration (82). D1 dopamine receptors may also regulate AMPA receptor surface expression. For example, an increase in the density of AMPA receptor surface clusters on nucleus accumbens neurons was observed after brief exposure to a D1 receptor agonist (83) (Figure 5⇓). This exciting finding suggests a new mechanism that may enable dopamine receptors—when stimulated during cocaine or amphetamine administration—to influence synaptic events underlying LTP and LTD. But, can drug-induced changes in LTP and LTD account for the persistence of addiction? Based on studies in the hippocampus, a model has been proposed in which LTP and LTD trigger a series of sequential changes that lead to alterations in the biochemical composition of the postsynaptic membrane, and ultimately to changes in the structure of dendritic spines (20). By tapping into LTP and LTD, drugs of abuse could influence these fundamental processes, accounting for the very long-lasting changes in brain structure and function that underlie addiction, which include changes in dendritic spines in the nucleus accumbens and prefrontal cortex (9,,10). It is also important to look beyond LTP and LTD, as drugs may produce neuroplasticity by regulating GABA-mediated inhibition in addition to glutamate-mediated excitation (77). Examining plasticity in neuronal circuits regulated by GABA is particularly important in light of recent work, which suggests that GABA-based drugs may be useful for treating addiction (84).
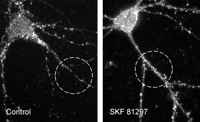
Dopamine regulates cell surface AMPA receptor expression. Insertion and removal of AMPA receptors from synaptic sites is important for LTP and LTD (16,17). In cultured nucleus accumbens neurons, D1 dopamine receptor stimulation increased the density of AMPA receptor puncta on the cell surface, suggesting a mechanism by which dopamine might influence LTP and LTD (83). Primary cultures were prepared from nucleus accumbens of postnatal day one rats. AMPA receptors on the cell surface were visualized by incubating live cells (twenty-one days in vitro) with antibody that recognizes the extracellular portion of the AMPA receptor subunit GluR1, fixing cultures, and then incubating with fluorescent secondary antibody. To quantify GluR1 surface expression, a standard length of neurite was defined using a circle 25 μM in diameter and the number of cell surface GluR1 puncta on a single neurite within the circle was counted using MetaMorph Imaging software. Left panel: Control conditions. Right panel: Incubation with the D1 agonist SKF 81297 (1 μM, for fifteen minutes) increased the density of surface GluR1 puncta. Nucleus accumbens interneurons are shown in these pictures, but the same results were obtained for medium spiny neurons, the projection neurons of the nucleus accumbens (83). In the same culture system, D1 receptor stimulation increased phosphorylation of GluR1 at the protein kinase A site, suggesting another way that dopamine may influence basic mechanisms of synaptic plasticity (81).
- © American Society for Pharmacology and Experimental Theraputics 2002
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵

Marina E. Wolf, PhD, is a Professor and is the acting-chair in the Department of Neuroscience at the Finch University of Health Sciences, Chicago Medical School. Please address correspondence to MEW. Email: marina.wolf{at}finchcms.edu; Fax (847)-578-3428

