CTLA-4: Acting at the Synapse
Successful immune cell control requires a delicate balance of positive and negative regulatory signals. Costimulatory pathways involving molecules such as CD28, inducible costimulator (ICOS), 4-1BB, and CD40L are essential coactivators of proliferation, cytokine production, and cell migration. To balance these signals, cell surface molecules like Fas, tumor necrosis factor-α receptor (TNFR), and programmed death-1 (PD-1) decrease T cell responses. Most prominent among these T cell regulators is cytotoxic T lymphocyte antigen-4 (CTLA-4), a homolog of CD28 that can interact with the CD28 ligands CD80 and CD86 (1). CTLA-4 inhibits T cell responses in a T cell receptor- (TCR)-dependent manner. For example, T cells treated with soluble CTLA-4 monoclonal antibody (mAb) have enhanced T cell proliferation and cytokine production, whereas surface-immobilized CTLA-4 mAb inhibits interleukin-2 (IL-2) production and cell cycle progression both in vitro and in vivo (2, 3). In addition, CTLA-4–deficient mice exhibit a CD4+ T cell lymphoproliferative disorder that leads to death of the animals a few weeks after birth (4, 5).
Several models have been proposed to explain the molecular basis for CTLA-4 inhibition (Figure 1A⇓). The first model proposes that CTLA-4 successfully competes against CD28 for CD80 or CD86 binding because CTLA-4 binds to them with much stronger avidity than does the CD28 costimulatory molecule (Ligand Competition Model) (6). Support for this model comes from studies showing that mutant CTLA-4 lacking a functional intracytoplasmic domain can suppress T cell function both in vitro (7), and in vivo (8). A second model suggests that CTLA-4 inhibits important downstream signaling pathways involving the extracellular signal-regulated kinase (ERK) and nuclear factor-kappa B (NF-κB) pathways (9, 10), subsequent to TCR-CD3 association and CD28 receptor engagement (Distal Blockade Model). In this model, CTLA-4-mediated inhibition depends on, but is downstream of, the TCR signal. Finally, a third model suggests that CTLA-4 associates with the immunological synapse and attenuates proximal signal initiated by the TCR (Proximal Blockade Model) (11, 12).
The immunological synapse is formed at the interface between the T cell and antigen-presenting cell (APC) membranes and is the hot spot for T cell activation. Upon T-cell–APC engagement, critical components of the TCR signal transduction machinery relocalize to this region initiating a biochemical cascade that leads to full T cell activation. In many instances, the interface forms highly organized scaffolds that create an immunological synapse (13, 14). Several early studies suggested that upon T-cell–APC engagement, CTLA-4 is exported to the cell surface at the site of cell–cell interaction, where CTLA-4 can regulate TCR-proximal signals (15, 16). These studies have been confirmed and extended by the work of Egen and Allison who demonstrate that CTLA-4 is at first localized in the uropod of activated T cells, where the microtubule organizing center (MTOC) is located (11). When T cells are restimulated with weak-agonist peptide-bearing APCs, CTLA-4–containing vesicles in the T cells rapidly relocalize just beneath the APC–T cell contact site. By comparison, stimulation with a strong agonist peptide induces translocation of CTLA-4 to the cell surface and association with the immunological synapse. At least two mechanisms regulate the cell surface expression of CTLA-4: tyrosine phosphorylation-dependent clathrin-mediated internalization (17–21), and the active release of the molecule from the intracellular compartment to the cell surface (22, 23). CTLA-4 has a tyrosine-based FVYVKM motif within its cytoplasmic tail that strongly interacts—in the dephosphorylated state—with the clathrin adaptor molecules AP1 and AP2. This interaction results in rapid internalization and trafficking of CTLA-4 to intracellular compartments (16–21). Many kinases can phosphorylate this tyrosine-based motif in vitro, and therefore prevent CTLA-4 from associating with clathrin and becoming internalized. The TCR-associated Src family kinases, Lck and Fyn, are the most likely tyrosine kinases involved, considering their localization within the immunological synapse (24, 25). This hypothesis is also consistent with observations showing that active translocation of CTLA-4 depends on TCR signal and is thus ZAP-70– and Lck-dependent (22), because Lck activation causes surface stabilization of CTLA-4 by direct phosphorylation of a regulatory tyrosine within the CTLA-4 tail (24, 25). Thus, the TCR signal might influence CTLA-4 expression by both translocation and stabilization, although whether this active transport depends on lysosome secretion or other mechanisms remains unclear (23).
CTLA-4 has been observed at the immunological synapse; however, there has been no direct biochemical basis to explain its inhibitory activity. CTLA-4 can directly associate with the TCRζ chain of the TCR–CD3 complex, and subsequent dephosphorylation of the ζ chain has been observed (12). These observations provided the basis for a model suggesting that the negative regulatory activity of this cell surface protein depends on the association of a tyrosine phosphatase because CTLA-4 lacks known intrinsic enzymatic activity. In this regard, two protein tyrosine phosphatases—Src homology 2 (SH2) domain–containing tyrosine phosphatase-1 (SHP-1) and SHP-2—can associate with the cytoplasmic tail of CTLA-4 (19, 26, 27); however, the role of these enzymes in the function of CTLA-4 is controversial because SHP-2 can positively regulate cell activities (28), and CTLA-4, in some cellular contexts, negatively regulates signals in the absence of SHP-1 (29).
PP2A, a protein serine-threonine phosphatase can associate with CTLA-4 (30). This protein phosphatase is known to negatively regulate the Ras-ERK pathway in some systems (31). This finding also fits with the localization of CTLA-4 to the immunological synapse, because Ras is activated within the synapse. However, PP2A also associates with CD28 (30), and is considered an activating molecule in T cell costimulation, possibly by dephosphorylating actin cytoskeleton–associated molecules (32). Thus, although CTLA-4 appears to effect early events in TCR signaling within the immunological synapse, the role of protein phosphatases in CTLA-4–mediated T cell inhibition remains unclear.
CTLA-4 appears to function at various stages of immune activation. CTLA-4–dependent blockade of TCR signaling effects the induction of tolerance following exposure to soluble antigens (33), reverses tolerance in experimental models of autoimmunity, and inhibits effector T cell function (34). In this regard, it is interesting to note that CTLA-4 is constitutively expressed on certain T cell subsets. Although naïve T cells do not express CTLA-4, memory CD4+ T cells do express low levels of the protein intracellularly (35). Moreover, both regulatory and tolerant T cells appear to express CTLA-4, and treatment of these cells with CTLA-4 mAbs long after antigen exposure can reverse tolerance. Thus, CTLA-4 might either inhibit T cell activation by increasing the threshold for productive T cell signaling activation, or attenuate ongoing immune responses by “putting the brakes” on the activation process.
During a primary T cell response to antigen presentation or during sustained T cell activation, CTLA-4 may participate more as an attenuator than as a threshold regulator because little CTLA-4 is expressed on the surface of these cells (Figure 1B⇓). This idea is supported by several recent findings. In one study, more CTLA-4 protein was expressed at the cell surface of proliferating T cells as compared to resting cells (36). Additionally, Egen and Allison showed that stimulation of T cells by a strong agonistic peptide caused the enhanced accumulation of CTLA-4 at the immunological synapse, where CTLA-4 inhibited further T cell activation (11).
By comparison, CTLA-4 is likely to increase the threshold for T cell activation under conditions where CTLA-4 is already present at the immunological synapse (Figure 1C⇓), for example, in recently activated memory T cells, tolerant T cells, regulatory T cells, or upon continued T cell stimulation. Once T cells are activated through antigen presentation, CTLA-4 becomes constitutively expressed, thus setting a signal threshold for secondary activation. In this case, the very small fraction of CTLA-4 molecules constitutively found on the surface of T cells blocks weak activation signals. Thus, the association of CTLA-4 at the immunological synapse in memory T cells or tolerant T cells may increase the signaling threshold for T cell activation by modulating either antigen-dependent (TCR) or -independent (CD28) signal transduction at the cell surface. The uncontrolled polyclonal expansion of autoreactive T cells in CTLA-4 knockout mice strongly suggests that CTLA-4 may increase the signaling threshold for activation to prevent the activation of T cells which have weak affinity to self ligands. In this context, the threshold will be set by the surface availability of CTLA-4, which is determined by the total amount and the spatial localization of CTLA-4 within one T cell.
When all the data are examined together, a picture unfolds suggesting that indeed the inhibitory function of CTLA-4 is not likely to be strictly a consequence of competition with CD28 for ligands, or inhibition of distal signaling cascades, but rather a very early event proximal to the signaling events following TCR engagement with antigen–major histocompatibility complexes (MHC). However, future studies of CTLA-4 regulation of T cell functions will be needed to gain better understanding of immune cell homeostasis as well as new insights for therapeutic manipulation.
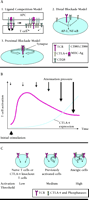
A. Three models for T cell inhibition by CTLA-4. 1. Ligand Competition Model wherein CTLA-4 can successfully compete with CD28 for CD80 and CD86 binding because of CTLA-4’s stronger avidity. 2. Distal Blockade Model wherein CTLA-4 inhibits distal pathways of TCR–CD28 signaling. 3. Proximal Blockade Model wherein CTLA-4 is recruited to the immunological synapse and blocks early signal transduction via the TCR. B. Attenuation model: In single T cells, CTLA-4 is synthesized during the time course of T cell activation and is recruited to the T cell surface. As a consequence, the T cell signal is attenuated and T cell activation is inhibited. C. Threshold model: In the absence of CTLA-4 (naïve cells and CTLA-4 knockout cells), the minimal requirement for T cell activation is very low. However, once CTLA-4 is induced (e.g., in previously activated cells or anergic cells), CTLA-4 is recruited to the immunological synapse where it increases the activation threshold either at the level of antigen specific (TCR) or co-stimulatory (CD28) signals.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Shunsuke Chikuma, PhD, is a Research Associate in the UCSF DiabetesCenter and is a member of the Bluestone laboratory.

Jeffrey A. Bluestone, PhD, is the A.W. and Mary Margaret Clausen Distinguished Professor and Director of the UCSF Diabetes Center; Professor in the Departments of Medicine, Microbiology and Immunology, and Pathology. Address comments to JAB. E-mail jbluest{at}diabetes.ucsf.edu; fax 415-564-5813.

