IRAK-4: A New Drug Target in Inflammation, Sepsis, and Autoimmunity
The cytokines interleukin-1 (IL-1) and IL-18 are important inflammatory mediators of such diseases as rheumatoid arthritis and inflammatory bowel disease. Their receptors, IL-1R–IL-1RAcP (IL-1R accessory protein) and IL-18R–IL-18RAcP, belong to a broader family of receptors that includes the Toll-like receptors (TLRs). This family is defined by a conserved cytosolic region termed the Toll-IL-1 receptor (TIR) domain. TLRs, crucially involved in the innate immune response to microbes, sense pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), peptidoglycan, dsRNA, and CpG DNA, which are recognized by TLR4, TLR2, TLR3, and TLR9, respectively. Additionally, TLR4 recognizes products of inflamed tissues such as the 60-kDa heat shock protein (HSP60).
Tremendous progress has recently been made in our understanding of how this family of receptors transmits signals. The TIR signaling system is attractive from a drug targeting point of view, given the role of IL-1 and IL-18 in inflammatory diseases, and the implication of TLRs in sepsis, autoimmunity (specifically TLR4 and TLR9), and possibly, atherosclerosis.
Recently, a key breakthrough has been made in the area of IL-1 and TLR signaling with the discovery of IRAK-4, which is recruited to receptor TIR domains and appears to be the initiating kinase in the signaling pathways activated following stimulation (1, 2). These receptors mediate cellular stimulation that culminates in the activation of well-known inflammatory signals such as the transcription factor NF-κB and stress-activated kinases, both of which are important drug targets with several inhibitors in Phase I clinical trials.
The TLR signaling pathways are strikingly complex. Ligand binding to the extracellular domain of TIR domain–containing receptors results in the recruitment of the soluble adapter molecule MyD88. The C-terminal TIR domain of MyD88 interacts with the receptor complexes, whereas the conserved death domain (DD) in its N terminus associates with the corresponding domains of IL-1R-associated kinases (IRAKs), homologs of the Drosophila protein kinase Pelle. Until recently, three mammalian members of this Ser-Thr kinase family were known: IRAK-1 (3), IRAK-2 (4), and IRAK-M (5). IRAKs characteristically contain a DD in their N terminus, and a central kinase domain followed by a unique 90 to 170-residue C-terminal domain. Under resting conditions, unphosphorylated IRAKs are bound to the Toll-interacting protein (Tollip) in the cytosol through a DD–DD interaction. In this cytosolic complex, IRAK is prevented from being phosphorylated prior to stimulation of the cell (6, 7). The binding of IL-1 to IL-1R leads to the recruitment of IL-1RAcP, which in turn transiently recruits the Tollip–IRAK complex resulting in activation. Furthermore, Tollip can directly associate with TLR2 or TLR4 (6). Kinetic studies suggest that the recruitment of MyD88 and Tollip–IRAK to the receptor occur independently (7). Moreover, no association between Tollip and MyD88 has been demonstrated, indicating that these proteins associate with distinct domains on the receptor (6). The initial, activating phosphorylation of IRAK at the receptor complex weakens its affinity for Tollip, consequently making it more accessible for interaction with MyD88 (7). Neither Tollip nor MyD88 are able to maintain interaction with fully activated, hyperphosphorylated IRAK-1, but can only associate with the unphosphorylated form (7, 8). Thus, the association of MyD88 with IRAK-1 is disrupted upon further phosphorylation of IRAK-1, which results in the release of IRAK-1 from the receptor complex. The successive aggregation of activated IRAK-1 with the soluble tumor necrosis factor-α (TNF-α) receptor–associated factor 6 (TRAF6) is the initial step for assembly of the signalosome and downstream activation of either NF-κB or stress-activated kinases.
The reported high turnover rate of IRAK-1 and Tollip can be ascribed to phosphorylation and subsequent ubiquitination (6). Rapid degradation of activated IRAK-1 ensures tight regulation of cytokine expression in response to IL-1R–TLR-mediated signaling. As opposed to IRAK-1, the IRAK-2 protein level remains constant after LPS treatment, which is an interesting indication of divergent roles of these homologs in the host defense against microbes (9).
IRAK-1 itself is an active kinase and undergoes autophosphorylation. However, overexpression of a catalytically inactive IRAK-1 mutant, and characterization of a kinase-inactive splice variant of IRAK-1 revealed that kinase activity is dispensable for IL-1 induced NF-κB activation (10, 11). Although IRAK-2 and IRAK-M both lack key catalytic residues, thus rendering them kinase-inactive, they nonetheless are both capable of signaling (4, 5). The only known heterologous substrate of IRAK-1 is Tollip; phosphorylation of Tollip in the activated receptor complex is thought to facilitate the dissociation of IRAK-1 from Tollip (6).
The nature of the IRAK-1–activating kinase at the receptor complex was, until recently, unknown. The fact that a heterologously overexpressed IRAK-1 kinase-negative mutant can still be phosphorylated in an IRAK-1–deficient cell line argues against self-activation (i.e., through autophosphorylation) (10). The recently discovered new member of the IRAK family, IRAK-4, appears to bridge the gap between the stimulated receptor complex and IRAK-1 activation. The kinase was discovered in a database search for IRAK homologs. It is the closest human homolog to the Drosophila Pelle, a key kinase in Toll signaling in Drosophila, with which it shares twenty-two percent amino acid sequence identity. IRAK-4 consists of the DD, a linker domain, and the kinase domain, but unlike the other IRAKs, IRAK-4 does not have a C-terminal extension (1). IRAK-4 knockout mice are resistant to LPS-induced septic shock, suggesting that the TLR4 signal pathway—known to be activated by LPS—was disrupted. Furthermore, the mice were resistant to shock induced by CpG DNA, peptidoglycan, and the dsRNA mimetic polyI:C, indicating that IRAK-4 is essential in TLR9, TLR2, and TLR3 signaling, respectively. In addition, the IRAK-4 knockout mice did not produce detectable amounts of TNF-α or IL-6 after IL-1 injection. This unresponsiveness to IL-1 stimulation was also observed in IRAK4−/− embryonic fibroblasts; however, full responsiveness to administered IL-1 was regained after stably transfecting IRAK-4 into these cells. Virus-infected IRAK-4–deficient mice were unable to produce interferon-γ (IFN-γ); because IFN-γ is produced in an IL-18–dependent manner after virus infection, signal transduction through the IL-18R also appears to be impaired. Activation of the mitogen-activated protein kinases (MAPKs) p38 and c-Jun N-terminal kinase by IL-1 or LPS is prevented in IRAK4−/− cells. Suzuki et al. furthermore demonstrated that IL-1 but not TNFα-induced NF-κB activity is dependent on IRAK-4 (1). Compared to the effects observed in IRAK-1 deficiency, the loss of IRAK-4 more severely affects TIR-mediated signaling (2). Partial redundancy of IRAK-1 with IRAK-2 and IRAK-M can explain this phenomenon; none of these IRAKs appear to functionally replace IRAK-4 in the knockout mice. For example, in contrast to the overexpression of IRAK-2 or IRAK-M, the overexpression of IRAK-4 in IRAK-1−/− cells does not activate NF-κB, suggesting that IRAK-4 might function upstream of IRAK-1. In addition, IRAK-4 appears to lie downstream of MyD88 and its homolog, the MyD88 adapter-like (Mal) protein, which has been implicated in TLR4 signals (12). IRAK-4 is the only IRAK that relies on its kinase activity for NF-κB activation—a kinase-negative variant of this protein acts as a dominant negative in IL-1 signal transduction, and prevents the IL-1-induced phosphorylation of IRAK-1. Most importantly, Li et al. demonstrated that IRAK-4 activates IRAK-1 by phosphorylating two crucial sites (Thr387 and Ser376) in the activation loop of IRAK-1 in vitro. Phosphorylation of IRAK-1 at these sites is critical for its kinase activity. IRAK-4 appears to associate transiently only with unmodified IRAK-1 upon IL-1 stimulation, suggesting that phosphorylated IRAK-1 is rapidly released as a modified substrate from its kinase (2).
The IRAK-4–catalyzed phosphorylation at the activation loop of IRAK-1 is presumably critical for the interaction of IRAK-1 with proteins like MyD88 and Tollip at the receptor complex. Once activated, IRAK-1 exhibits autophosphorylation and leaves the receptor complex in its hyperphosphorylated form to further associate with TRAF6.
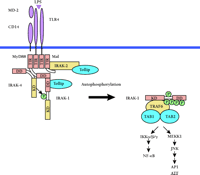
Molecular interactions at the TLR4 signaling complex. Upon stimulation with LPS, IRAK and Tollip are recruited to the receptor complex comprising TLR4, MD-2, and CD14. Simultaneous binding of IRAK-1 and its upstream kinase IRAK-4 to MyD88 ensures efficient activating phosphorylation of IRAK-1, which is released from the complex after further autophosphorylation. An additional adapter, Mal, recruits IRAK-2 to the complex. See text for details. TIR, Toll-IL-1 receptor domain; DD, death domain; KD, kinase domain.
Additional insights into the events at the activated IL-1R and TLRs have been gained in a study on MyD88s, an alternatively spliced form of MyD88 (13). This truncated variant lacks the intermediate domain connecting the N-terminal DD with the C-terminal TIR domain. Its expression is carefully regulated, and it appears to be a natural repressor for IL-1 and LPS signaling by preventing IRAK-1 phosphorylation. However, IRAK-1 can still interact with MyD88s, but it does not become phosphorylated (13). These observations imply that the MyD88 interdomain region, missing in MyD88s, is required for IRAK-1 phosphorylation possibly through the recruitment of IRAK-4. Simultaneous binding to distinct domains of MyD88 would bring IRAK-4 and its substrate IRAK-1 in close proximity and facilitate IRAK-1 phosphorylation. Thus, MyD88 could play an important role as a scaffolding protein in IL-1R–TLR signaling. A scheme describing TIR-dependent signaling is shown for TLR4 (Figure 1).
IRAK-4 is therefore the most receptor-proximal kinase so far described for IL-1, IL-18 and TLR signaling. Inhibiting its kinase activity would limit the pro-inflammatory effects of IL-1 and IL-18, and might also be beneficial in blocking chronic harmful TLR-dependent signaling in such conditions as sepsis or in autoimmunity.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Luke A. J. O’Neill, PhD, is an Associate Professor of Biochemistry and head of the Inflammation Research Group in the Department of Biochemistry, Trinity College, Dublin 2, Ireland.
Claudia Wietek, PhD, is a postdoctoral research fellow in the Department of Biochemistry, Trinity College, Dublin 2, Ireland. Address correspondence to CW. E-mail wietec{at}tcd.ie; fax ++33 1 677 2400.

