Molecular Targets for Therapeutic Intervention after Spinal Cord Injury
Abstract
In an effort to develop therapies for promoting neurological recovery after spinal cord injury, much work has been done to identify the cellular and molecular factors that control axonal regeneration within the injured central nervous system. This review summarizes the current understanding of a number of the elements within the spinal cord environment that inhibit axonal growth and outlines the factors that influence the neuron’s ability to regenerate its axon after injury. Recent insights in these areas have identified important molecular pathways that are potential targets for therapeutic intervention, raising hope for victims of spinal cord injury.
INTRODUCTION
Few survivable injuries compare with the devastating physical and psychological consequences of the paralysis that results from trauma to the spinal cord. Each year, over 10,000 North Americans, most below the age of thirty, sustain such spinal cord injury (1). Whereas the loss suffered by each of these individuals is incalculable, societal costs, in terms of the medical, surgical, and rehabilitative care for patients with acute and chronic spinal cord injuries, come to four billion dollars per annum (estimated from decade-old data) (2). Clearly, therapies to enhance neurological function after spinal cord injury are urgently needed. This need has sparked great interest in the neuroscience community, and many exciting experimental strategies have emerged. The purpose of this review is to summarize some of these advances, with a particular focus on the molecular elements deemed to play an important role in preventing axonal regeneration after spinal cord injury, and how these elements can be targeted by therapeutic interventions.
INJURY CONSIDERATIONS: PRIMARY AND SECONDARY DAMAGE
Typical injury to the human spinal cord occurs during blunt, non-penetrating trauma (e.g., motor vehicle and diving accidents), whereby the bony and ligamentous components of the spinal column are displaced and impart sudden mechanical forces to the spinal cord. A number of animal (predominantly rodent) injury models have been developed in an attempt to reproduce such injuries (3). The primary, mechanical disruption of tissue is quickly followed by a complex cascade of secondary injury processes that include regional blood flow alterations, electrolyte homeostasis perturbations, free radical generation, excitotoxicity, inflammation, and apoptotic cell death (4, 5). These secondary mechanisms damage spinal cord tissue that was otherwise spared during the initial insult and thus represent a potential target for neuroprotective strategies. In current clinical practice, the mainstay of neuroprotective therapy is limited to the judicious medical resuscitation and stabilization of the acutely traumatized patient (6) and high-dose administration of the steroid methylprednisolone (7–10). A number of other pharmacological means of influencing the course of secondary injury are currently in various stages of animal and human evaluation (5, 11). Novel neuroprotective agents will undoubtedly play an important role in the future for minimizing neurological damage in the acutely injured patient.
As secondary injury processes subside during the weeks and months following the trauma, the site of spinal cord injury becomes typically characterized by disrupted axons, a cystic cavity encased within a gliotic scar, and variable amounts of intact tissue surrounding the lesion (12, 13). In this peripheral rim of intact tissue reside axons that are either uninjured or that have lost part of their myelin sheaths. The clinical challenge at this stage is to promote compensatory sprouting of the remaining intact axons or the re-growth of severed axons across the injury site (Figure1⇓). Permanent paralysis after spinal cord injury testifies to the fact that this challenge has gone largely unanswered. It has become apparent that axonal regeneration after spinal cord injury fails both because of elements within the injury environment that inhibit axonal growth, and because the central nervous system (CNS) neurons themselves demonstrate a relatively weak intrinsic ability to regenerate axons after injury. Although many exciting experimental approaches have been developed to overcome these obstacles individually, it is generally agreed that an optimal therapeutic strategy for human patients will require combinatorial treatments that address both extrinsic and intrinsic barriers to regeneration.
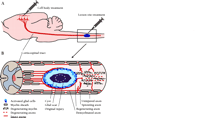
Experimental treatment of rat spinal cord injuries. A. Treatments are applied typically at the lesion site but can also be applied to the cell body. B. Cellular events typify a chronic spinal cord injury. Primary cellular damage spreads the insult to surrounding cells and creates secondary damage that can supersede the original injury. Secondary damage includes a central acellular fluid-filled cyst surrounded by a glial scar. In a typical crush injury, axons per se are often left uninjured, intact but partially demyelinated, or severed. Degeneration of axons and myelin occurs distal to the injury site. Treatment paradigms may promote regeneration of severed axons into or around the injury site, possibly into the distal spinal cord. In addition, treatments often promote the sprouting of uninjured axons into areas of deafferentation.
INHIBITORY ELEMENTS WITHIN THE ENVIRONMENT OF THE INJURED CNS
Although the inhibitory environment of the CNS has been recognized for nearly a century (14), not until the past two decades have CNS myelin and the glial scar become appreciated as primary blockades to CNS regeneration after injury (15–17). Much has been done to identify the specific elements of CNS myelin and the glial scar that inhibit axonal growth, and recent advances in our understanding of the intracellular signalling pathways that mediate these inhibitory effects may offer potential therapeutic targets.
Myelin Inhibitors of Axonal Regeneration
In the late 1980s, pioneering work in the laboratory of Martin Schwab demonstrated that oligodendrocytes and their myelin membranes are major inhibitors of axonal growth within the CNS (18, 19). Corroborating this finding, John Steeves and colleagues demonstrated that the failure of axonal regeneration within chick spinal cord coincide with the onset of spinal myelination; in fact, the experimental delay of myelination acts to extend the permissive period for axonal regrowth (20). Subsequently, at least five separate inhibitory elements have been attributed to myelin: arretin (21); a chondroitin sulfate proteoglycan (22); the relatively well-characterized Nogo-A protein (23–25); myelin associated glycoprotein (MAG) (26, 27); and the recently identified oligodendrocyte myelin glycoprotein (OMgp) (28).
Nogo-A
Schwab and colleagues provided some of the earliest evidence that axonal growth could be promoted by targeting specific aspects of CNS myelin (18). They biochemically separated 35- and 250-kDa inhibitory fractions from CNS myelin and developed a monoclonal murine antibody known as IN-1 that could block their inhibitory properties in vitro (29). Subsequent in vivo application of the monoclonal antibody resulted in substantial axonal sprouting and some long-distance corticospinal axonal regeneration within the adult mammalian CNS (30). Importantly, this antibody treatment was associated with improved performance in a variety of functional tests, including open-field locomotion, rope climbing, and food pellet reaching (31, 32). Although the partial spinal cord injury models used in these early antibody studies could conceivably have permitted functional recovery due either to regeneration of injured axons or to sprouting and compensation from uninjured axons (Figure 1⇑), subsequent work has suggested that much of the benefit derived from the antibody treatment is in fact mediated by sprouting and functional plasticity of spared axons (33). With the hopes of translating this experimental approach to the clinical setting, a humanized version of the monoclonal IN-1 antibody has been generated and shown to be beneficial in animal models of partial spinal cord injury (34) and stroke (35).
The IN-1 antibody recognizing several components of myelin was also instrumental in the characterization and cloning of the target antigen (23–25, 36), the neurite outgrowth inhibitor now known as Nogo. The cloned gene encodes three integral membrane proteins (Nogo-A, -B, and -C) and has been assigned (on the basis of sequence homology to three previously sequenced proteins) to the reticulon family of proteins. Consistent with its role as an inhibitor within CNS myelin, Nogo-A is highly expressed by oligodendrocytes but not by Schwann cells (23). The original descriptions of the Nogo protein have led to some uncertainty regarding the actual components responsible for axonal inhibition (37). Whereas the Schwab and Walsh laboratories ascribe the inhibitory effect to the N-terminal domain of Nogo-A (24, 25), the Strittmatter group has assigned inhibitory functionality to an extracellular loop of sixty-six amino acids (Nogo-66) found in all three Nogo isoforms (23).
Fortunately, significant progress has been made in the recent identification and cloning of a receptor for the Nogo-66 protein (NgR) (38). This landmark study demonstrates that the NgR is anchored to the cell membrane through a glycophosphatidylinositol linkage, suggesting that NgR is in fact part of a receptor complex. The role of the NgR in axonal growth inhibition is illustrated by the positive correlation between its expression level in chick neurons of the dorsal root ganglion (DRG) and responsiveness to myelin (i.e., Nogo) inhibition. Furthermore, transfection techniques that cause (normally unresponsive) E7 retinal ganglion cells to express NgR confer sensitivity to Nogo-66. Interestingly, although Nogo-66 inhibits neurite outgrowth of E12 chick DRG neurons, the soluble form of the N-terminal domain is not inhibitory, lending further credence that the sixty-six–residue extracellular domain possesses “Nogo” activity (38).
In experiments to delineate the residues of Nogo-66 that are specifically responsible for inhibitory activity, Strittmatter and colleagues found that a truncated peptide comprising amino acids 1 to 40 in the Nogo extracellular polypeptide loop (NEP1–40) fails to inhibit neurite growth, and in fact acts as a competitive antagonist of Nogo-66 binding to the NgR (39). After dorsal hemisection of the rat thoracic spinal cord, local application of NEP1-40 significantly increases the number of corticospinal and serotonergic axon profiles distal to the site of injury, presumably indicative of antagonism of the NgR and resultant neuronal sprouting/regeneration. However, models of partial injury, such as dorsal hemisection, leave room for uncertainty as to whether the newly sprouted axons arise from injured axons in the dorsal column versus ininjured ventral axons. Nevertheless, many corticospinal axon sprouts occur in the dorsal spinal cord, suggesting origination from the injured dorsal corticospinal tract. Perhaps most excitingly, the injured animals treated with NEP1-40 show significant and sustained (i.e., at twenty-eight days post-injury) improvement in open field locomotor testing, which the authors attribute to both early sprouting and subsequent long-distance regeneration (39). Clearly, NEP1-40 raises the exciting potential for specifically targeting myelin inhibition of axonal regeneration in the context of human spinal cord injury, and may find relevance in the treatment of neurotraumatic and neurodegenerative disorders.
Myelin Associated Glycoprotein (MAG)
Although NEP1–40 is clearly effective as an antagonist of Nogo-66, its ability to promote neuronal regeneration is, even at high concentration in vitro, incomplete (39). Elements other than Nogo that reside within CNS myelin and inhibit neuronal regeneration must thus be considered. One such element is myelin-associated glycoprotein (MAG), which inhibits neurite outgrowth from adult CNS neurons in vitro (26, 27). The role of MAG as an inhibitor of regeneration in the peripheral nervous system is clear from the increased axonal growth of peripheral nerves manifested in certain mice that are deficient in MAG (40). Within the CNS, however, the importance of MAG as an impediment to axonal regeneration has been difficult to establish, most likely because of the presence of the other inhibitors of axonal growth (41).
Our understanding of the function of MAG would be significantly enhanced by the identification of its receptor, and quite recently, several molecules have emerged as candidate receptors for MAG: the gangliosides GD1a and GT1b (43); the p75 neurotrophin receptor (44); and interestingly, the Nogo-66 receptor, NgR (45, 46) (Figure 2⇓). The indication that the NgR might mediate the inhibitory effect of both Nogo and MAG (and OMgp, as discussed below) is particularly intriguing, because such a receptor would represent a point of convergence for numerous therapeutic strategies (Figure 2⇓). The Filbin group (45) has recently demonstrated that the removal of GPI-linked proteins such as the NgR from the surface of cerebellar neurons precludes their interaction with an experimental variant of MAG (i.e., MAG–Fc), and as a result, neurite outgrowth is no longer inhibited. The interaction between MAG and NgR is more directly demonstrated by the binding of MAG to NgR-expressing CHO cells, as well as by the binding of a soluble NgR to MAG-expressing CHO cells. Moreover, the in vitro inhibition of neurite outgrowth by MAG can be overcome by three methods: with a polyclonal antibody to NgR; with a soluble version of NgR to compete for MAG binding with membrane-bound NgR; and with a recombinant dominant-negative version of the NgR (N-NgR). Interestingly, the binding of the experimental variant of MAG to NgR-expressing CHO cells is inhibited by soluble form of Nogo-66, implying that the two proteins compete for the same binding site on the NgR (45). The Strittmatter group has provided similar evidence regarding the relationship between MAG and the NgR, but the authors state that competitive inhibition between MAG and Nogo-66 was not observed (46). Also, the NEP1-40 peptide, which competitively antagonizes the inhibitory effect of Nogo-66, had no effect on the inhibition of neurite growth by MAG, suggesting that the Nogo-66 and MAG bind to NgR at distinct sites, a proposition supported by the lack of homologies between the two proteins (46).
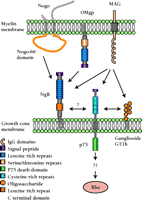
Inhibitory myelin components and their putative receptors. Three myelin membrane proteins that inhibit axonal growth are Nogo, OMgp, and MAG. All three may bind to the Nogo receptor (NgR) in the axonal growth cone, although downstream signaling events are less clear. MAG has also been shown to bind both to ganglioside GT1b and to the p75 low-affinity neurotrophin receptor. p75 may regulate the affects of MAG through Rho-GTPase signaling to the actin cytoskeleton. (OMgp, oligodendrocyte-myelin glycoprotein; MAG, myelin associated glycoprotein; see text for details.).
Oligodendrocyte Myelin Glycoprotein (OMgp)
The list of identified inhibitory elements of CNS myelin was recently lengthened to include oligodendrocyte-myelin glycoprotein (OMgp) (28), a 105-kDa GPI-linked protein described over a decade ago to be highly expressed in CNS myelin (47). Quite remarkably, like Nogo-66 and MAG, OMgp also appears to act through the NgR. Wang et al. identified OMgp as a potential culprit in the growth cone–collapsing activity of the proteins released by the treatment of myelin with phosphatidylinositol-specific phospholipase C, and demonstrated that the expression of recombinant OMgp in COS-7 cells induces the collapse of E13 chick DRG growth cones (28). The screening of a COS cell cDNA expression library with OMgp fused to alkaline phosphatase revealed NgR as a possible receptor for OMgp, findings that were subsequently confirmed in co-precipitation studies. Most compelling, NgR transfection of E7 DRG neurons (normally not yet expressing the NgR) rendered these neurons sensitive to Omgp—a gain of function similarly elicited with respect to Nogo-66 (38). Deletion constructs of the NgR revealed that OMgp and Nogo-66 bind to overlapping regions of the receptor, and consistent with this finding, no additive effect was observed with regard to growth cone collapse when OMgp and Nogo-66 were administered coincidentally as opposed to single administration of either factor.
Hence, NgR has recently emerged as a single receptor for the three major myelin proteins known to inhibit axonal outgrowth: Nogo-A, MAG, and OMgp (Figure 2⇑). These exciting findings point strongly to the convergence of numerous inhibitory elements onto the same signaling pathway (see below), which may represent a specific molecular target for interventions to overcome the failure of CNS regeneration.
Glial Scar Inhibitors of Axonal Regeneration
At the site of CNS injury, a complex cellular reaction represented by the activation of astrocytes, microglial cells, and the invasion of macrophages, fibroblasts, and meningeal cells gives rise to what is commonly referred to as the “glial scar” (15, 48–51). Although most spinal cord injuries are caused by non-penetrating trauma, potentially important differences in the nature of glial scarring exist after blunt versus penetrating injury, with a connective tissue basal laminar component and meningeal cell invasion being more prominent in the latter. The glial scar is generally accepted to represent an impediment to axonal regeneration after CNS injury, but the relative significance of its molecular and cellular constituents in inhibiting axonal growth is not entirely clear. Nevertheless, due to their expression of inhibitory molecules (see below) and the potentially impenetrable physical barrier created by the dense intertwining of their processes, reactive astrocytes are widely believed to play an important role in inhibiting nerve regeneration (49). The extent to which this inhibition is due to the physical environment created by the tightly interwoven astrocytic processes is uncertain, although there is some evidence that the three dimensional structure erected by these astrocytes has some implications for axonal growth. For example, whereas astrocyte monolayers support DRG neurite outgrowth in vitro, outgrowth is not observed within astrocytes packed into a three dimensional tube structure (52).
The importance of astrocytes as in vivo inhibitors of CNS axonal growth is implied in a transgenic mouse model where the ablation of astrocytes results in pronounced axonal sprouting penetrating across the CNS injury (53). Perhaps the most convincing demonstration that glial scars inhibit axonal regeneration comes from Silver and colleagues, who show that embryonic or adult DRG cells, microinjected into the spinal cords of young adult rats without trauma, can extend axons for long distances along myelinated white matter tracts until they encounter the glial scar (16). The long-distance regeneration is prevented if the microinjection inadvertently induces local glial scarring at the transplant site, thus entrapping the DRG axons. Reactive astrocytes (50, 54–56) and surrounding inflammatory cells and oligodendrocyte progenitors (57, 58) express inhibitory chondroitin sulfate proteoglycans (CSPGs), which have lately achieved some notoriety as possible targets for therapeutic intervention. Two recent studies have demonstrated the use of chondroitinase ABC (Ch-ABC) to degrade CSPG in vivo and thereby induce axonal growth (59, 60). In fact, intrathecal bolus infusions of Ch-ABC, at the time of dorsal column crush lesion and every other day for ten days thereafter, results in a rostro-caudal 4-mm span of spinal cord that is free of glycosaminoglycan side chains (59). Anterograde axonal tracing of sensory fibers, furthermore, reveals some growth along the wall of the injury cavity, and corticospinal fibers 1 and 2 mm proximal, as well as 1 mm distal, to the lesion epicenter show significant growth. Ch-ABC treated animals also manifest, upon electrophysiological stimulation of the cortex, a small short latency wave 2 mm caudal to the lesion (indicating the propagation of postsynaptic potentials across the injury site) that can be abolished by a subsequent re-lesion to the dorsal column. Finally, the Ch-ABC–treated animals show significant behavioral improvements (albeit transient in some cases) in tape removal, beam walking, and footprint analyses.
Although the results of Ch-ABC–treated animals are all very promising results, the partial nature of the dorsal column injury used in these studies most likely spares ventral and dorsolateral corticospinal tracts, making it difficult to determine whether the increase in corticospinal profiles observed distal to the injury site represents growth from axotomized neurons or collateral sprouts from spared corticospinal fibers (Figure1). Increased terminal arborization from such sprouts could also increase postsynaptic field potentials and account for the observed electrophysiological findings. The inhibitory properties of CSPG are sometimes associated primarily with its core protein (22, 61), so that some inhibitory activity would be resistant to Ch-ABC treatment. As the researchers of the study demonstrate, CSPGs are also expressed by oligodendrocytes (62), and thus, Ch-ABC treatment may promote corticospinal plasticity normally suppressed by oligodendrocytes/myelin. Even so, the promotion of axonal sprouting of spared fibers and subsequent functional recovery clearly has significant clinical relevance, as the majority of human spinal cord injuries (even with functionally “complete” paralysis) are in fact anatomically partial injuries. The distinction between strategies that promote axonal regeneration versus sprouting would, however, be important for those with anatomically complete lesions (for example, full spinal cord transections from penetrating trauma), in which axonal sprouting could be crucial. Despite many unanswered questions, the promising results from the use of Ch-ABC suggest that the approach of targeting inhibitory elements within the glial scar holds promise for clinically appreciable treatments of spinal cord injury.
Although much attention has been placed on CSPGs, it should be mentioned that several other putative axonal growth inhibitors have been ascribed to the glial scar, or to its extracellular connective tissue components, such as collagen-IV (63), tenascin (64, 65), class-3-semaphorins (66, 67), and Eph3B (68). The relative importance of these molecules as inhibitors of axonal growth is still a matter of some debate, awaiting conclusive loss-of-function verification in vivo. The digestion of basal laminar collagen is reported to promote some regeneration in the retrospenial fornix (63), but the inability to reproduce these findings in the spinal cord setting (69) leaves uncertainty around the candidacy of collagen as a target of molecular intervention.
INTRINSIC REGENERATIVE ABILITY OF INJURED CNS NEURONS
That the permissive environment of peripheral nerves allows neurons from the CNS to extend their axons after injury was demonstrated by Tello and Ramon y Cajal almost a century ago (14), and has been reiterated with more contemporary methods in landmark work performed by Richardson, Aguayo, and colleagues (70). The regenerative limits of mature CNS neurons, however, lie in stark contrast to the fairly robust regenerative abilities of neurons projecting into the peripheral nervous system. This contrast is certainly explained in part by the different environments encountered by neurons of the central as opposed to the peripheral nervous system (the latter environment being far more conducive), but it is also clear that CNS neurons intrinsically fail to mount or to maintain some of the gene expression programs necessary for a regenerative response to injury (71, 72). The regenerative response is thought to involve the expression of genes that encode a wide spectrum of proteins, including transcription factors, cytoskeletal proteins, growth cone proteins, and cell adhesion molecules (73). The identification and clinical manipulation of the expression of these proteins represents a potential therapeutic strategy for neuron regeneration and spinal cord repair (74). It is important to recognize that our current appreciation of the full battery of genes that are regulated to produce axonal outgrowth is fairly rudimentary, although microarray technology will undoubtedly accelerate our understanding of such regulation in the future.
Lessons from the Dorsal Root Ganglion: GAP-43 and Conditioning Lesions
The sensory neurons of the DRG, which possess axons that project into the central and peripheral nervous systems, have provided an extremely useful model for studying intrinsic neuronal regenerative propensity. Richardson and Issa made the intriguing observation in 1984 that injured central sensory axons regenerate into permissive peripheral nerve transplants inserted into the dorsal columns much more readily if a “conditioning” injury to the peripheral sensory axon is implemented beforehand (75). These observations suggested that the peripheral axotomy initiated a molecular response within the cell body that was necessary for the regeneration of the subsequently injured central axon. It was later shown that such a conditioning peripheral lesion could augment the intrinsic regenerative response sufficiently to drive injured central axons across the otherwise nonpermissive spinal cord injury environment (76). One of the components that may play an important role in driving this regenerative response is GAP-43, a protein localized to the growth cone where, by virtue of its interactions with calmodulin, PI(4,5)P2, G proteins, and the actin cytoskeleton, it influences axonal growth and synaptic plasticity (77, 78). The correlation between increased GAP-43 expression and axonal growth and sprouting (79, 80) has made GAP-43 a commonly used indicator of neuronal growth propensity, although one should be aware that there do exist examples in which GAP-43 is neither sufficient (81) nor necessary (82) for axonal growth. Bomze at al. have demonstrated in vivo that the concomitant overexpression of both GAP-43 and CAP-23 (a related growth cone protein) results in significant regeneration of the central DRG axons into peripheral nerve transplants, whereas overexpression of either protein alone failed to promote such axonal regeneration (83). Still, the most extensive regeneration was observed after a conditioning peripheral axotomy, indicating that the two genes (i.e., those encoding GAP-43 and CAP-23) do not account completely for the conditioning effect of the peripheral nerve lesion.
Cyclic AMP as an Intrinsic Determinant of Growth Propensity
The DRG model has also been extremely valuable for the identification of cAMP as an important intrinsic determinant of axonal growth. Two very recent studies, in particular, have strongly implicated cAMP in the promotion of DRG central axon regeneration (84, 85), much like the increased growth propensity observed with a conditioning peripheral axotomy (76). In studies by Neumann et al. and Qiu et al., a single injection of a cAMP analog into DRG neurons in vivo significantly enhanced their neurite outgrowth in vitro from short, highly branched neurites to an elongated pattern with few branches. The cAMP injection was also associated with regenerative sprouting in vivo of ascending injured sensory axons after a dorsal column spinal cord lesion, with axonal growth occurring mainly along the wall of the injury site cavity (84, 85). The extent to which the cAMP-activated regenerating sensory axons can extend beyond the injury site and make synaptic contact with target neurons is difficult to determine. Consistent with the enhanced regenerative propensity achieved with a conditioning lesion to the peripheral branch of the DRG neuron, cAMP levels were also found to be significantly increased by peripheral axotomy.
Marie Filbin’s group has presented data that may explain some of the mechanisms by which cAMP mediates these growth-inducing effects. In particular, the cAMP-enhanced growth that allows DRG neurons to extend neurites on inhibitory substrates (86) is dependent on activation of cAMP-responsive binding protein (CREB), which results in the upregulation of arginase I and subsequent downstream activation of polyamine synthesis (87, 88). The implication of polyamine synthesis in DRG regeneration is consistent with the increase in ornithine decarboxylase activity (a rate-limiting enzyme in polyamine synthesis) observed in regenerating facial motoneurons after axotomy (89); indeed, exogenous application of polyamines stimulates such regeneration (90).
It is worth noting that in the recent demonstrations of in vivo regeneration of DRG axons within the spinal cord, the cAMP was administered to the DRGs two (85) or seven days (84) before the spinal cord injury was actually induced. Obviously, within the context of translating these important findings into a clinically relevant strategy for spinal cord repair, the beneficial effects of manipulating cAMP levels would have to be demonstrated after injury. Nevertheless, these and other studies have provided important insights into the role of cAMP in neuronal regenerative capacity.
Neurotrophic Factor Enhancement of Regenerative Competence
Neurotrophic factors are small proteins that play diverse roles in cell survival, axonal regeneration, and synaptic plasticity, and may thus find therapeutic application in spinal cord injury and a host of other neurological disorders (91). The implication of cAMP (and downstream PKA activation) in neuronal regenerative capacity suggests that DRG neurons, “primed” with neurotrophic factors that may function via cAMP could extend neurites even on nonpermissive substrates such as MAG and myelin (86). Indeed, the in vivo treatment with brain-derived neurotrophic factor (BDNF) or glia-derived neurotrophic factor (GDNF) promotes regeneration of sensory neurons across the dorsal root entry zone and into the spinal cord, with demonstrable functional recovery (92). We have similarly found that BDNF, applied to the cell bodies of acutely and chronically injured rubrospinal neurons, can enhance regenerative capacity as evidenced by the upregulation GAP-43 and Tα1 tubulin, as well as axonal growth into peripheral nerve transplants (93, 94). Also, application of BDNF to corticospinal neurons promotes the sprouting response within the spinal cord (95). Although a direct link between neurotrophic factors and cyclic nucleotides was not evaluated in our studies of the rubrospinal and corticospinal system, it is certainly conceivable that the application BDNF to the neuronal cell body stimulates the activation of the MAPK/ERK pathway, with subsequent activation of CREB, as has been previously demonstrated for hippocampal neurons in vitro (96) and in vivo (97). Hence, the activation of CREB may represent a point of convergence for the intracellular effects of neurotrophic factors and cAMP, mediating common downstream changes in the expression of relevant genes such as arginase I and possibly many others that have not yet been identified.
Increasing intracellular levels of cAMP with phosphodiesterase inhibitors such as rolipram represents a potentially useful strategy for promoting axonal growth, and may be pharmacologically easier to achieve than the application of neurotrophic factors, although overcoming undesirable side effect (e.g., nausea) may be important considerations in both cases. Although the growth-inducing effects of neurotrophins and cAMP discussed herein involve molecular alterations at the level of the cell bodies, we should point out that cAMP has also been shown to play an important role at the level of the growth cone, influencing the growth cone’s response to axonal guidance cues (discussed below).
MANIPULATING GROWTH CONE SIGNALING TO PROMOTE REGENERATION—THE ROAD TO RHO
The actual extension of an axon occurs at its distal tip, where the growth cone plays a key role in axonal regeneration by negotiating the environment and integrating signals from an array of guidance molecules. The growth cone cytoskeleton is primarily composed of filamentous actin (F-actin) in its peripheral and central domains, and microtubules extending from the central domain along the entire shaft of the axon (reviewed in 98–100). The peripheral domain comprises two dynamic F-actin–based structures: filopodia, made from polarized bundles of F-actin; and lamellipodia, made from a meshwork of F-actin (Figure 3A⇓). These actin-rich structures contribute to the force necessary for the forward extension of the growth cone (reviewed in 101, 102); thus, a better understanding of how actin dynamics are regulated could provide key insights into how axonal regeneration might be promoted. It turns out that the members of the Rho family of small guanosine triphosphatases (GTPases), in particular RhoA, Rac1, and Cdc42 (reviewed in 103), appear to play a critical role in the control and regulation of actin dynamics in the growth cone and thus have emerged as potentially important molecular targets for inducing axonal regeneration.
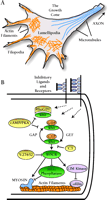
The growth cone: Features and inhibitory signaling pathways. A. A growth cone has distinct actin filament-based features, including distal, finger-like filopodia and central, fan-shaped lamellipodia. B. Schematic diagram of the Rho-signaling pathway in the growth cone, a possible pathway of convergence for numerous extracellular stimuli. Receptor activation somehow alters the balance of Rho-GEF and Rho-GAP activity so as to favor an increase in activated, GTP-bound Rho. Conversely, Rho activity can be down-regulated by RhoGDI sequestration to the cytoplasm, an effect promoted by intracellular cAMP/PKA elevation. Rho-GTP activates its downstream effector kinases, of which ROCK is depicted here. ROCK in turn has effects on several substrates that affect actin events that lead to growth cone advance or retraction. (GAP, GTPase activating proteins; GEF, guanosine nucleotide exchange factors; RhoGDI, Rho guanosine nucleotide dissociation inhibitor; ROCK, Rho-kinase; PKA, protein kinase A.)
In neuronal cell lines, Rac and Cdc42 promote the formation of lamellipodia and filopodia, whereas Rho activity causes neurite retraction and growth cone collapse (104–107). [In primary neuron cultures, however, the effects of Rho GTPases are not so straightforward, indicating that an optimal level of Rac and Cdc42 GTPase activity, or optimal cycling between the GTP- and GDP-bound forms, is needed for effective neurite outgrowth (108–110).] Activated (i.e., GTP-bound) Rho modulates the activities of numerous proteins, among which Rho-kinase/ROKα/ROCK-2 (ROCK) is the best characterized. ROCK mediates Rho signaling to the actin cytoskeleton (111) by phosphorylating myosin light chain (MLC) (112, 113); ROCK also phosphorylates LIM-kinase (114, 115), which is thereby activated so that it, in turn, phosphorylates cofilin (116), which thereby contributes, along with phosphorylation of myelin light chain, to growth cone retraction (Figure 3B⇑).
A number of studies have demonstrated a prominent role of the Rho-ROCK pathway in mediating the growth cone collapse and/or retraction effects of several inhibitory molecules found within the injured CNS. Inhibition of Rho by C3 exoenzyme promotes axonal elongation from chick DRG cells that are otherwise inhibited by Semaphorin 3A (117). In addition, C3 exoenzyme–treatment (or the inhibition of ROCK with the specific ROCK inhibitor Y-27632) promotes axonal elongation of retinal ganglion cell axons that are otherwise inhibited by EphA receptor signaling (118). Rho inhibition by C3 exoenzyme–treatment thus overcomes the inhibitory effects of myelin, as well as of purified MAG on the neurite (117, 119). Importantly, the in vivo application of C3 exoenzyme promotes axonal regeneration of crushed optic nerves in adult rats by as much as 500 μm from the crush site (119). This C3–exoenzyme treatment has been reported to promote axonal regeneration of the corticospinal tract after a full spinal cord transection injury (120), and we eagerly await the full-length publication of these results. Consistent with these findings, our laboratory has observed that the extension of DRG axons grown on aggrecan, an inhibitory CSPG substrate, could be stimulated by up to tenfold when ROCK was inhibited by Y-27632 (J. Borisoff et al., unpublished results).
As an interesting adjunct to the discussion of cAMP and its role in neuronal regenerative propensity, elevating cAMP levels may be yet another method of inhibiting Rho, in perhaps a somewhat less specific manner than the C3 exotoxin. Rho can undergo phosphorylation in a PKA-dependent manner (121), resulting in its inactivation and the prevention of stress fiber formation in non-neuronal cells. Experiments using rat brain membrane fractions indicate that this PKA-dependent phosphorylation inhibits Rho by enhancing its interactions with the Rho-GDP-dissociation inhibitor (122). Interestingly, cAMP and PKA activation have been shown in vitro to be critical for the directional growth responses of Xenopus spinal neurons to chemoattractive and chemorepulsive agents such as BDNF and MAG (123, 124).
If Rho and its subsequent activation of ROCK represent a key pathway that mediates axonal inhibition, and myelin and its elements are prominent effectors of similar axonal inhibition, what are the molecular means by which these intra- and extracellular components interact? Very recent work has provided significant insight into this question. Inhibition of neurite growth due to MAG is mediated by the binding of MAG to the p75 neurotrophin receptor, with subsequent activation of Rho (44). The relationship among MAG, p75, and Rho is further supported by observations that MAG colocalizes with and binds to p75, activates RhoA, and fails to inhibit adult DRG neurons that express nonfunctional p75 receptors (44). Because MAG also binds to the NgR (46), one could also speculate that the p75 receptor interacts with the GPI-linked NgR to form part of the elusive signal transducing NgR receptor complex (Figure 2⇑).
In summary, it is well established that members of the Rho family of small GTPases plays a central role in mediating signaling from permissive as well as inhibitory extracellular guidance molecules. With regard to the numerous inhibitory molecules that have been identified as potential obstacles to axonal regeneration, there is accumulating evidence that their intracellular signaling pathways converge by means of Rho, making the Rho pathway a potentially important target for repair strategies (e.g., ROCK inhibition). Although the mechanism by which cAMP influences extension and directional decisions of the growth cone have not been firmly established, it is tempting to speculate that it is at least in part also mediated through Rho inactivation (Figure 3B⇑). In this regard, a therapeutic intervention that increases levels of cAMP and PKA activation might achieve dual functionality by exerting rapid effects on the growth cone cytoskeleton (conceivably through Rho) and relatively long-term effects, through CREB activation of gene expression, that augment the intrinsic neuronal growth propensity.
CONCLUSIONS
Many significant advances have been made recently in our understanding of some of the important molecular mechanisms involved in promoting and inhibiting axonal regeneration within the CNS after injury. With the recognition that the injured spinal cord is enormously complex, and that attempting to treat each of its numerous interrelated elements individually would be daunting, the identification of key pathways upon which many of these elements converge is crucial to therapeutic strategies. Much work remains to be done in moving these exciting discoveries forward, because some of them (e.g., the demonstration that the pre-injury administration of cAMP promotes axonal growth within the injured spinal cord), although undisputedly important, represent proof of principle only. Nevertheless, progress that has been made even within the last year bodes well for the future development of therapies.
Acknowledgments
The authors gratefully acknowledge the funding of the Neuroscience Canada Foundation (NCF), Canadian Institutes for Health Research (CIHR), Rick Hansen Institute, and the National Science and Engineering Research Council of Canada and the Christopher Reeve Paralysis Foundation. BKK is the Gowan and Michele Guest NCF/CIHR Research Fellow. WT is the Rick Hansen Man in Motion Chair in Spinal Cord Research at the University of British Columbia.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Brian K. Kwon, MD, (center), completed clinical training in orthopedic surgery at the University of British Columbia before arriving at ICORD as a Gowan and Michele Guest Fellow, where he is in his final year of studies for his PhD in Neuroscience. Jaime F. Borisoff (right) is presently completing his PhD studies in Neuroscience at the University of British Columbia, where he is supported by a Rick Hansen Studentship. Wolfram Tetzlaff, MD, PhD, (left) obtained his medical degree in Germany, trained at the Max-Planck-Insitute for Psychiatry in Munich, and obtained his PhD in Neuroscience at the University of Calgary. Subsequent to faculty positions at the University of Calgary and Ottawa he now holds the Rick Hansen Man in Motion Chair in Spinal Cord Research at UBC and is Associate Director for Discovery Science for ICORD.

