p53 Moves Into Control of Cell Morphology
Loss-of-function mutations in the p53 tumor suppressor protein contribute to aberrant regulation of cell cycle progression and aberrant apoptosis in approximately 50% of tumors (1). In tumors where p53 is not mutated, p53 can be destabilized by overexpression of MDM2 (murine double minute 2), which promotes its ubiquitination and degradation, or by loss of p14ARF, which antagonizes MDM2 function (2). p53 is a transcription factor that coordinates the cellular response to genotoxic stresses by promoting the transcription of genes involved in cell cycle arrest, DNA repair, and apoptosis (3) (Figure 1⇓). In response to cell stress or DNA damage, the ability of p53 to regulate transcription is increased by a number of mechanisms including phosphorylation and acetylation (4). A multicellular organism is therefore protected from the effects of genetic damage either by p53-mediated cell cycle arrest allowing the damaged DNA to be repaired, or by p53-mediated apoptosis leading to the elimination of damaged cells. In addition to the loss of cell cycle checkpoints and the inability to correctly regulate apoptosis, tumor cells undergo many characteristic morphological changes; these include the loss of polarity, altered cell–cell and cell–substratum contacts. An interesting publication by Gadéa et al. demonstrates an unexpected role for p53 in regulation of these processes (5). This raises the intriguing possibility that altered control of cell cycle, DNA repair, and apoptosis may not be the only ways loss of p53 contributes to tumorigenesis.
Cell morphology is controlled by the Rho family of small guanosine triphosphatases (GTPases) that include Cdc42, Rac1, and RhoA. When bound to GTP, these proteins recruit a range of target proteins that regulate the cytoskeleton (6). Cdc42 regulates the formation of actin-rich microspikes (also called filopodia) that can initiate cell movement and cell contacts with the extracellular matrix; microspikes often precede the formation of larger Rac1-dependent membrane protrusions called lamellipodia that drive the motility of many cell types (7) (Figure 2⇓). Activation of RhoA leads to the formation of actin stress fibers that generate contractile forces and are responsible for retracting the tails of migrating cells. Rho family proteins have also been implicated in the regulation of transcription and cell proliferation (8). There is increasing evidence that Rho proteins become deregulated by over-expression in tumors, and in some cases this correlates with progression of the disease (9). Gadéa et al. now demonstrate that overexpression of p53 prevents Cdc42 from promoting the formation of microspikes and regulating the orientation of cells undergoing directional cell movement (Figure 2⇓). In addition, activation of p53 by etoposide, adriamycin, or TNFα prevents the formation of Cdc42-dependent microspikes in response to bradykinin. The function of Rac1 and RhoA was not affected by overexpression of p53. Analyses of two p53 mutants demonstrated that the ability to block Cdc42-dependent processes correlated with the ability of p53 to bind DNA and to function as a tumor suppressor. Furthermore, mouse embryo fibroblasts lacking p53 constitutively exhibit microspikes and enhanced spreading on fibronectin—both processes that are promoted by active Cdc42. These data suggest that loss of p53 may promote cell motility in some contexts. Indeed, Alexandrova et al. found that restoring p53 expression in fibroblasts lacking p53 reduces cellular motility in a wound healing assay (10), and Gadéa et al. mention that overexpression of p53 antagonizes fibroblast motility in a similar assay.
The work of Gadéa et al. raises several questions that warrant further investigation. Cdc42 can activate the mitogen-activated protein kinases (MAPKs) Jun N-terminal kinase (JNK), and p38, resulting in changes in transcription (8), and it will be interesting to determine if this aspect of Cdc42 function is enhanced by loss of p53. In addition, p53 is a member of a family of transcription factors (11); it is unclear whether other family members share the ability to suppress the effects of active Cdc42. In the light of this study, it is tempting to speculate that increased Cdc42-driven motility of tumor cells with p53 mutations may contribute to the poor prognosis associated with p53 mutation in some tumor types (12–14). There may also be a role for the connection between p53-dependent and Cdc42-dependent processes outside the context of tumor biology. For example, p53-induced inhibition of Cdc42 may stop the movement of migrating cells, enabling cellular damage to be repaired before migration continues. In addition, the interference of Cdc42 function during p53-driven apoptosis may contribute the morphological changes associated with apoptosis (15).
The mechanism by which p53 affects Cdc42 function remains an interesting question; Gadéa et al. show that Cdc42 is not directly activated by loss of p53 and that overexpression of p53 blocks the effects of a constitutively active Cdc42 mutant. These data suggest that activation of p53 must block microspike formation downstream of Cdc42. It is likely that p53 mediates its effects by controlling the transcription of genes involved in responding to active Cdc42. The formation of microspikes is dependent upon the interaction of Cdc42 with the Myotonic Dystrophy-Related Cdc42-binding Kinases (MRCKs) and the structural proteins WASP and N-WASP (16–18), whereas the orientation of the Golgi apparatus is dependent on Cdc42 binding to a complex containing atypical members of the protein kinase C family (19). It will be interesting to test if loss of p53 promotes the expression or activity of these molecules: several other genes involved in cytoskeletal regulation, such as α-actin, are known transcriptional targets of p53 (20, 21). However, it is not clear if changes in actin levels are involved in altering the ability of Cdc42 to control microspike formation and orientation of the Golgi apparatus. DNA microarray analysis of p53-regulated genes may help elucidate the mechanism by which p53 counteracts Cdc42 function.
The high frequency of p53 mutations in human cancer has led to extensive research into strategies to modulate p53 function. Pharmacological agents have been developed that can restore the function of mutant p53 to wild-type activity and can retard tumor growth in mouse models (22, 23). An inhibitor of p53 function has also been generated and the development of these compounds may counteract the side-effects of current genotoxic therapies used in cancer treatment (24). The identification of this new link between p53 and Cdc42 has several implications for therapies related to p53: firstly, the effect of p53-targeted drugs on the cytoskeleton and cell motility needs to be investigated. Secondly, if loss of p53 leads to changes in cell morphology, then the molecules that regulate these changes may be good drug targets. Indeed, the Rho family of proteins and their binding partners are already being investigated as potential targets for cancer therapy. Compounds that block the geranyl-geranylation of Rho proteins can reduce tumor growth in animal models (25, 26), and inhibition of the Rho-associated coiled-coil kinase (ROCK), a kinase whose activity is regulated by RhoA, prevents the dissemination of tumor cells in vivo (27). The identification of the p53-specific genes that mediate the interference with Cdc42 function may also suggest new therapeutic targets.
The work of Gadéa and colleagues has elucidated a new aspect to p53 function that may relate to its function as a tumor suppressor gene; understanding how p53 modulates the regulation of cell morphology by Cdc42 will be an exciting challenge. Thus, insights gained from these studies will most likely lead to the identification of new targets for cancer therapy.
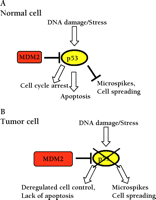
p53 functions as a tumor suppressor gene. A. In unstressed wild-type cells, the activity of p53 is relatively low, and its degradation is promoted by MDM2. Genotoxic stress leads to the activation of p53, resulting in cell cycle arrest or apoptosis, and a reduction in Cdc42-dependent microspikes. B. In tumor cells p53 is often mutated, or degraded owing to increased MDM2 activity. This leads to deregulated cell proliferation, failure to undergo apoptosis, and an increase in Cdc42-dependent functions. MDM2, murine double minute 2.
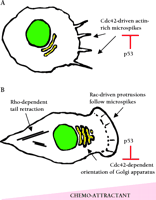
Role of Rho proteins in cell motility. A. Chemotactic stimuli can promote Cdc42-dependent microspikes that can be suppressed by activation of p53. Microspikes can promote the formation of contacts between the cell and the substratum and cell spreading. B . Microspikes precede the formation of larger Rac1-dependent membrane protrusions that drive cell movement. Cdc42 activity is required to orient the Golgi apparatus in the direction of movement; the activation of p53 prevents this process. RhoA promotes the generation of the contractile force that is required to move the remaining part of the cell behind the protrusion.
Acknowledgments
The advice of Mathew Coleman, Steven Hooper, and Chris Marshall was much appreciated. E.S. is funded by Cancer Research UK.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Erik Sahai, PhD, is currently a post-doctoral fellow with Chris Marshall at The Institute of Cancer Research in London. He has a long-standing interest in the role of Rho proteins in tumorigenesis; his current work focuses on interactions between Ras and Rho protein signaling and how deregulation of Rho proteins contributes to the altered morphology and increased motility of tumor cells. Previously, his doctoral studies with Richard Treisman focused on the role of the ROCK family of kinases in Rho-mediated transformation. E-mail erik{at}icr.ac.uk

