Molecular Genetics of Human Cognition
Abstract
An intimate relationship exists between our capacity for learning and memory and the domains of human cognition. This relationship becomes all too clear when a genetic disruption causing the diminished capacity for memory formation results in an individual with a pronounced cognitive deficit. Only recently, through the identification of human genetic mutations that result in mental retardation and learning disorders, have we begun to gain some insight into the genetic and molecular basis of human cognition. The following review describes some of the genetic causes and biochemical consequences of several recently identified genetic disorders that result in a disruption of memory formation and subsequent decline in human cognitive function.

Introduction
Cognition can be defined as the active intellectual processes through which information is obtained, transformed, stored, retrieved, and used by the brain. The ability to form long-lasting memories and to retrieve these memories when necessary is the axis upon which human cognitive ability revolves. Memory formation not only defines who we are as a species in our capacity for learning, but our personal memories and experiences help to define who we are as individuals. It should be recognized that the capacity for this basic, intrinsic ability to form new memories is ultimately genetic in origin. This becomes extremely evident when a genetic aberration causes a disruption in the capacity for memory formation and manifests in an individual as a pronounced cognitive deficit.
We have reached a point where clinical and basic science research have converged and provide us insight into the molecular basis of human cognition. Only recently have neuroscience researchers begun to solve any of the mysteries surrounding proteins and signaling pathways that underlie the formation of lasting memory in the mammalian brain. In parallel, clinical research has identified many genes responsible for human learning disorders. Thus, we are at a point where new hypotheses can be formulated concerning the molecular mechanisms that underlie human mental retardation syndromes. Additionally, the identification of specific genes and their products in human disorders that are associated with mental retardation has greatly facilitated our understanding the basic mechanisms involved in the formation of human memories.
The following review examines the genetic causes and biochemical consequences of several recently identified disorders that result in a disruption of human cognitive ability. Although our understanding of many of these disorders is incomplete and still under intense investigation, we discuss the implications of the emerging molecular basis of these disorders on our current understanding of learning and memory mechanisms. One focus of our review will be how new, genetically engineered mouse models of human mental retardation syndromes have given us insight into the molecular biology of human learning disorders.
Memory Formation: A Molecular Primer
Much progress has been made in the last decade toward understanding the molecular blueprint for long-term memory formation in the mammalian brain. Long-term memory formation begins at the plasma membrane of neurons, at synapses located on dendritic spines. High frequency presynaptic activity at these sites leads to the release of glutamate, which activates a host of glutamate receptors on the postsynaptic spine (Figure 1⇓). Glutamate receptors are generally divided into two broad categories: ionotropic and metabotropic. Ionotropic receptors are ligand-gated channels that can depolarize the membrane [i.e., α -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate subtypes] or allow calcium influx [i.e., N -methyl-d -aspartate (NMDA) receptors]. Metabotropic receptors are coupled to intracellular second messenger cascades. High-frequency stimulation results in a large postsynaptic depolarization, which allows for activation of the NMDA-subtype of ionotropic glutamate receptors leading to Ca2+ influx into the spine. Moreover, the attendant high levels of glutamate allow for activation of metabotropic glutamate receptors, which are coupled to the phospholipase C (PLC) pathway. The increase in intracellular Ca2+ and the activation of metabotropic glutamate receptors in the spine leads to activation of a number of postsynaptic protein-kinase signaling pathways, including the Ca2+ -calmodulin kinase II (CaMKII), cAMP-dependent protein kinase (PKA), protein kinase C (PKC) pathways, and the Ras pathway. One downstream target that is activated by all of these pathways is the mitogen-activated protein-kinase (MAPK) termed extracellular-regulated kinase (ERK). Once stimulated, ERK phosphorylates and activates ribosomal S6-kinase 2 (RSK2), which in turn phosphorylates the transcription factor cAMP response-element (CRE) binding protein (CREB). CREB recruits a number of transcriptional coactivators, including CREB binding protein (CBP), and initiates a wave of transcription that is vital for the consolidation of long-term memory. Newly synthesized transcripts must be transported out of the nucleus and translated into functional proteins, which effect lasting changes in synaptic strength by altering the electrical properties of the membrane, increasing the responsiveness to neurotransmitter, and even changing the number and size of synapses.
The formation of memory, thus, depends upon the coordination of numerous cellular processes. A consequence of the complexity of memory formation however, is that disruption of one, or a few of the molecular steps involved can be deleterious. For example, deficits in human memory formation (i.e., mental retardation) are caused by specific defects in the molecular pathways responsible for memory formation. In the sections that follow, we will explore the molecular basis of some molecular defects along this pathway that result in cognitive deficits in humans.
Cell Signaling: Defects in Information-Flow from the Membrane to the Nucleus
Recent work has shown that a number of signaling cascades must be activated to successfully form long-term memories. In retrospect, it is not surprising then, that a number of deficits in human cognition arise from derangements in the functioning of these signaling pathways vital to the formation of memory.
Angelman Syndrome
Angelman Syndrome (AS) is a severe form of mental retardation that occurs in one of every 15,000–20,000 births. AS is usually accompanied by epilepsy, a puppet-like gait, dysmorphic facial features, a happy disposition with bouts of inappropriate laughter, hyperactivity, sleep disorders, and lack of speech (1, 2) .
In 1997, the gene responsible for AS was identified as the ubiquitin-protein ligase, Ube3a (3, 4) . An unusual characteristic of this defect is that the Ube3a locus undergoes imprinting, whereby only the maternal allele is active in specific tissues. The paternal silencing of the Ube3a gene occurs in a brain region-specific manner; the maternal allele is active almost exclusively in the hippocampus and cerebellum (5) . The most common genetic defect leading to AS is an ∼4 Mb maternal deletion in chromosomal region 15q11–13 causing an absence of Ube3a expression in the maternally imprinted brain regions. Ube3a codes for an E6-AP ubiquitin ligase, an enzyme involved in the ubiquitin-proteasome pathway. Ubiquitination targets proteins for degradation through the proteasome pathway. However, E6-AP ubiquitin-ligase chooses its substrates very selectively and the four identified E6-AP substrates have shed little light on the possible molecular mechanisms underlying the human AS mental retardation state. The development of mouse models for AS has increased our understanding of the genetic alterations and physiological consequences underlying this disorder.
There are two mouse models for AS which replicate the abnormalities and brain-specific imprinting of Ube3a observed in humans (3, 6, 7) . The mouse model developed by Jiang et al. has provided a variety of information regarding the neural basis of the cognitive defects caused by AS (7) . Initial studies of mice that do not express maternal Ube3a showed severe impairments in hippocampal memory formation (7) . Most notably, there is a deficit in a learning paradigm that involves hippocampus-dependent contextual fear conditioning. In addition, maintenance of long-term synaptic plasticity in hippocampal area CA1 in vitro is disrupted in Ube3a−/− mice (7) . Thus, current research using this mouse model focuses on the molecules required for hippocampal synaptic plasticity. The results from murine gene-knockout studies provide a link between hippocampal synaptic plasticity in vitro and formation of hippocampus-dependent memory in vivo, and suggest that the expression of synaptic plasticity, as measured by LTP formation, underlies human hippocampus-dependent memory formation.
Neurofibromatosis
One of the most common single-gene disorders causing cognitive disorders in humans is Neurofibromatosis type 1 (NF1), which affects approximately one in 4,000 individuals. The predominant phenotypic characteristic of NF1 is the development of benign neurofibromas of the peripheral nervous system. Other clinical manifestations include malignant tumors (gliomas or malignant peripheral nerve sheath tumors), skin discoloration, and skeletal dysplasia. NF1 leads to varying levels of mild cognitive impairment in 40–65% of patients (8) . The heterogeneity of NF1 gene mutations is thought to contribute to its lack of complete penetrance for various phenotypes, including cognitive disruption (9) .
The product of the NF1 gene, neurofibromin, is highly expressed in brain during embryogenesis, and in adults it is primarily expressed in pyramidal and Purkinje neurons, Schwann cells, and oligodendrocytes. NF1 mRNA undergoes alternative splicing resulting in four different protein isoforms (type I-IV); however, the predominant neuronal forms are produced through alternative splicing of exon 23a, resulting in type I (NF1-I, exclusion of exon 23a) or type II (NF1-II, inclusion of exon 23a) protein (10) .
Exon 23a contributes to the regulation of GAP [guanosine triphosphatase (GTPase) Activating Protein] activity intrinsic to NF1 (Figure 2⇓). This domain (including the GAP) is important for interactions of NF1 with Ras (10) , a low molecular weight GTPase that acts as a critical relay in signal transduction by cycling between an active conformational state (Ras•GTP) and an inactive state (Ras•GDP) (Figure 2A⇓). The GAP activity of NF1 promotes the hydrolysis of GTP and the formation of Ras•GDP, thus inhibiting Ras activity. Guanine nucleotide exchange factors (GEFs) counterbalance the effects of GAP proteins by catalyzing the exchange of GDP for GTP, leading to increased amounts of Ras•GTP. Therefore, loss of NF1-dependent inhibition of Ras could lead to the learning deficits seen in NF1 (11) .
In a series of elegant studies, Costa et al. investigated the role of Ras in the cognitive disruption of NF1 (12) . NF1 heterozygous knockout animals, which show a deficit in spatial learning, were crossed with animals deficient in Ras activity. Remarkably, the genetic diminishment of Ras activity was sufficient to rescue the spatial learning deficit observed in the NF1 heterozygous mice. Additionally, learning deficits were rescued in NF1-heterozygous animals treated with a farnesyltransferase inhibitor, suggwsting that hyperactivation of the Ras pathway is the likely cause of learning deficiencies in NF1-deficient mice and patients with NF1.
The question remains: what are the downstream targets of Ras hyperactivation that lead to cognitive disruption? Ras regulates the Raf–ERK cascade in a variety of cells, including hippocampal neurons (Figure 2B⇓). Therefore, it is likely that the ultimate consequence of hyperactivation of the Ras signal transduction cascade lies in the dysregulation of one or more ERK-regulated transcription factors, including CREB, Elk-1 and c-Myc.
Coffin-Lowry Syndrome
Coffin-Lowry Syndrome (CLS) is an X chromosome-linked disorder. Affected males with CLS are characterized by an abnormal gait, facial abnormalities (protruding forehead, wide noses, and irregular teeth), skeletal abnormalities (osteopenia, short stature, and muscular hypodevelopment) and severe mental retardation (13, 14) . As with other X-linked disorders, heterozygous females exhibit similar but much less severe characteristics. Before the development of reliable genetic testing for the disease, CLS was often misdiagnosed because of the variability in its manifestation, which mimics many other X-linked mental retardation conditions.
CLS is caused by a disruption of the gene encoding the 90-kDa r ibosomal S 6 serine-threonine k inase-2 (Rsk2 ), located at Xp22.2. Early studies of the Rsk2 gene and analyses of Rsk2 mutations from CLS patients have revealed that a wide variety of mutations of the Rsk2 gene—including splice alterations, missense, frame-shift and nonsense mutations—are responsible for CLS (15) . Initially identified as a ribosomal S6-kinase, Rsk2 also phosphorylates and activates a number of nuclear proteins including CREB (15, 16) . In the CNS, the ERK signaling pathway activates of Rsk2, which is responsible for PKA-dependent CREB activation in the hippocampus (Figure 2B⇓).
Dufresne et al. have produced a viable, Coffin-Lowry mouse model through the disruption of Rsk2 (17) . Rsk2 mutant mice exhibit deficits in motor learning and coordination, and have severe deficits in spatial learning. However, much of what can be inferred about the role of Rsk2 in cognitive function has come from studying the activity of ERK1, ERK2, and CREB in synaptic plasticity and memory formation. The requirement for the ERK and CREB in synaptic plasticity and formation and consolidation of long-term memories is well established. Thus, molecular components of memory formation up- and downstream of Rsk2 are involved in mammalian cognition. Yet, many questions still exist about the role(s) of Rsk2 in neuronal function and the mental aberrations seen in CLS. For example, evidence implicates Rsk2-mediated phosphorylation of SOS, a GEF for Ras, as a possible mechanism for negative-feedback regulation of the Ras–MEK–MAPK pathway (18) . Further investigation of the mechanisms underlying deficits in synaptic plasticity and memory formation in CLS-model mice will be necessary.
Cognitive Disruption at the Nucleus
Changes in cellular physiology due to the direct action of various second messengers and signaling pathways are short-lived at best. Long-term changes in cell function are usually effected through changes in expression of one or more genes. Thus, ultimately gene expression is necessary for the integration of external signals with long-term internal cellular function. It is not surprising that many diseases of cognition are due to derangement of nuclear function.
Down Syndrome: Aberrant Gene Dosage
First described by John Langdon Down in 1866, Down Syndrome (DS) is the most common form of mental retardation. DS occurs in one out of 800 live births, and accounts for nearly 30% of moderate to severe mental retardation. There are a number of stereotypical physical attributes associated with DS including short neck, short broad hands, flat nasal bridge, and narrow palate. IQs of DS patients range from 40–70, depending on the severity of the illness. Individuals with DS display a pronounced delay in the development of language, and exhibit deficits in various forms of short-term and spatial memory. There is an increased risk for early development of Alzheimer dementia, which usually occurs around age thirty-five.
The molecular basis for DS is the presence of an extra copy of chromosome 21, referred to as trisomy 21 (Ts21). Chromosome 21 is 45 Mb long and contains 303 genes. A region of ∼5 Mb of chromosome 21 has been implicated in the development of mental retardation in DS patients (19) , and within this critical region lie several genes that may affect memory formation (20) . Overexpression of genes from this locus is thought to interfere with their normal regulation and to lead to the cognitive deficit in DS (21) . Two mouse models with trisomy have been generated (Ts16, Ts65Dn), both of which mimic most of the DS human phenotype including severe memory impairments (20) . In the following sections we will briefly review some of the more prominent genes in this region.
Perhaps the most notorious gene the critical region of chromosome 21 encodes is the amyloid precursor protein (APP). The APP-derived peptide known as amyloid beta peptide (Aβ ) is one of the primary components of the plaques found in the brains of Alzheimer’s disease patients (22) . Mice that overexpress Aβ have deficits in LTP induction and memory formation (23) . Recently, Dineley et al. (24) showed that Aβ can activate the ERK signaling cascade through α 7-nicotinic acetylcholine receptors. Moreover, Aβ -dependent dysregulation of ERK signals leads to aberrant increases in phosphorylation of CREB (24) . As mentioned above, the ERK–CREB signaling pathway is essential in the formation of long-term memories in a number of different organisms.
Another gene present on chromosome 21 implicated in memory formation is the Cu2+ /Zn2+ superoxide dismutase (SOD1 ) gene. Superoxides can potentiate synaptic transmission and are required for induction of hippocampal LTP in vitro (25, 26) . In mouse models, overexpression of SOD, as would occur in DS, leads to decreases in ambient levels of superoxide and impairs induction of hippocampal LTP in vitro and hippocampus-dependent long-term memory formation in vivo (27) .
DYRK1A, the human homolog of the Drosophila Minibrain kinase, is also well studied. DYRK1A is located in the DS-critical region of chromosome 21 and is expressed in several structures of the adult CNS including the cortex, hippocampus and cerebellum (28) . Dyrk1A expression is significantly elevated in the CNS during development in DS humans and in a mouse model for DS, Ts65Dn (29) . Transgenic mice that overexpress Dyrk1A (TgDyrk1A) display several behavioral abnormalities that are similar to those seen in DS. Specifically, TgDyrk1A mice are delayed in their acquisition of mature locomotor skills and have deficits in spatial learning (30) .
Human Ts21 neurons appear to have significantly different passive electrical properties from normal diploid neurons (20) . Ts21 neurons are more excitable at rest, and show dramatically different rates of action potential depolarization and repolarization (20) . Neurons isolated from Ts16 mice, the other mouse model for DS, also have abnormal, passive electrical properties; however, the differences observed do not exactly replicate those found in the human disease (20) . Not surprisingly, there are several genes on human chromosome 21 involved in the regulation of neuronal excitability, including the inward-rectifying K+ channels Kcnj6/Kir3.2, Kcnj15/Kir4.2, and Kcne1 (20) .
Rett Syndrome
Andreas Rett, an Austrian pediatrician, first described Rett syndrome (RS) in 1966 (31) . In the United States, RS did not gain major attention until 1983, when it was further characterized by Hagberg et al. (32) . RS is an inherited, X-linked disease that afflicts about one in 15,000 females by ages 2–18 years of age, and is estimated to be the second leading cause of mental retardation in women, behind DS (33) . Development during the first six months of life is normal in RS patients, with symptoms appearing between three months to three years. The trademark of RS is display of continuous, stereotypical hand movements, such as wringing, washing, clapping or patting, which appear after the loss of purposeful hand movement. Other signs of RS include decreased growth (including microcephaly), abnormal respiration, gait ataxia, autism, seizures and other neurologic dysfunctions. Several mapping studies narrowed the location of the putative RS locus to Xq28 (34) , and in a percentage of patients, a mutation of the methyl CpG binding protein 2 (MeCP2), whose gene is located in Xq28, is correlated with Rett syndrome (35) .
MeCP2 functionally connects DNA methylation to gene silencing (Figure 3⇓). DNA methyltransferases catalyze the methylation of cytosine at position 5 using S -adenosylmethionine as a methyl donor. Methylation usually occurs at CpG (cytosine, phosphorylated guanine) dinucleotides. The distribution of CpG in the genome is not random; most CpG is found in repeating sequences and in CpG-enriched areas (islands). Interestingly, CpG islands are most commonly found in the promoter region of genes. Once methylated, 5mCpG is bound by MeCP2, which then recruits a complex of proteins including histone deacetylases and transcriptional corepressors such as Sin3A (36) . Ultimately, the histones associated with the 5mCpG become hypoacetylated, promoting tight association between the DNA and the histones and resulting the formation of a stable, transcriptionally-repressive chromatin complex.
The role that MeCP2 might play in the memory deficits observed in RS is still unclear. DNA methylation is involved in genome imprinting and dosage-compensation. Therefore, MeCP2 would be predicted to play a prominent role during development. Indeed, early attempts to create mice lacking MeCP2 resulted in embryonic lethality (37) . However, RS is a progressive disease that does not result in symptoms until early childhood. More recent attempts to create MeCP2-deficient mice have succeeded (38, 39) . These mice display some of the characteristics of RS including reduced brain and body size, tremors, hypoactivity, and irregular gait (38, 39). However, there are no studies detailing the cognitive performance of these mice.
Another strain of mouse has been developed that more closely approximates the mutations commonly found in RS patients (40) . In this mouse model, the last one-third of MeCP2’s protein-coding region was removed (MeCP2308/y ) (40) . MeCP2308/y mice share phenotypic similarities with RS including seizures, impaired motor coordination, hypoactivity, increased anxiety, and display some repetitive forelimb movement (40) . However, MeCP2308/y mice have no memory deficits, suggesting that either MeCP2 does not account for the entire RS phenotype, or that this particular MeCP2 mutation does not exactly replicate all the cognitive aspects of RS.
Rubinstein-Taybi Syndrome
Described by Rubinstein and Taybi in 1963 (41) , Rubinstein-Taybi Syndrome (RTS) occurs approximately once in every 125,000 live births, and accounts for one in 300 patients with mental retardation. There are several physical traits associated with RTS including altered facial features, broad digits, and blunted growth (42) . RTS is an inherited, autosomal dominant disease. Genetic studies have determined that the locus responsible for RTS maps to chromosome 16p13.3, which contains the gene for the transcriptional coactivator CREB-binding protein (CBP) (43) . Subsequent studies have shown that RTS patients have a variety of mutations in CBP, including point mutations and 5’- or 3’-deletions (44) . It should be noted that patients are typically heterozygous for a mutation in CBP, suggesting that RTS is due to either haploinsufficiency or a dominant-negative effect (43) . Homozygous mutations of CBP are likely embryonic lethal in humans. Homozygous CBP-deficient mutant mice die in the early embryonic stage (E10.5–E12.5), apparently as a result of massive hemorrhage caused by defective blood vessel formation in the CNS, and exhibit developmental abnormalities as well as delays in both primitive and definitive hematopoiesis.
CBP is important in linking CREB activation to gene transcription. The transcription factor CREB lies dormant in the nucleus until activated by phosphorylation. Once activated, phospho-CREB (P-CREB) recruits CBP and binds to CREs within the DNA (Figure 4⇓). CBP is thought to acetylate histones associated with the CRE, disrupting the chromatin within the promoter and facilitating the binding of the RNA polymerase II holoenzyme (45) . Therefore, an increase in CREB-mediated transcription is associated with an increase in histone acetylation, especially histones located near CREs. Indeed, several studies indicate that histone acetylation accompanies increases in CREB-dependent transcription (45) .
CREB plays an important role in consolidation of long-term memory. If CBP is vital for CREB-mediated transcriptional regulation, it follows that CBP would also be important for long-term memory formation. Therefore, mutations in CBP or disruption of CBP expression would be predicted to lead to impairments in memory formation, such as those seen in RTS. Two different strains of mice with mutated CBP alleles have been studied (46, 47) . The first strain, which contains a null mutation leading to haploinsufficiency, had no memory impairments (46) . However, the second strain, which was heterozygous for a dominant-negative mutant form of CBP that possessed only the CREB binding domain (CBPDN ), mimicked nearly all aspects of RTS (47) . Specifically, CBPDN mice had altered facial features and stunted growth (47) . Moreover, CBPDN mice had dramatic deficits in long-term memory formation, with normal short-term memory (47) , suggesting that RTS is caused by a dominant-negative mutation in one allele of CBP. Furthermore, these results demonstrate that CBP plays a critical role in the consolidation of long-term memory. The histone-acetyl transferase (HAT) activity possessed by CBP, was absent from the CBPDN mice, might specifically underlie the deficits in consolidation of long-term memory. If the HAT activity is indeed the critical factor underlying RTS, then this suggests that histone deacetylase inhibitors might be able to treat this disease.
Myotonic Dystrophy
Muscular dystrophy describes a class of diseases whose ultimate pathology is striated muscle wasting and eventual death. The most common form of muscular dystrophy, myotonic dystrophy (DM1) occurs once in every 8,000 live births (48) . Symptoms usually begin to appear in adulthood, and include mild mental retardation, myotonia, abnormal cardiac conduction, hypersomnia, cataracts, insulin-dependent diabetes, and testicular atrophy, and premature balding in men. Congenital cases of DM1 are more pronounced than cases of DM2, and characteristics include severe mental retardation, hypotonia, facial dysplegia, inability to feed and respiratory problems. We will focus on the molecular basis of the cognitive impairments associated with DM1.
DM1 is an inherited, autosomal dominant disease that has been mapped to chromosome 19q13.2–13.3 (49) . Subsequent studies identified DM1-associated protein kinase (DMPK ) as the gene mutated in DM1. The exact mutation responsible for DM1 was determined to be an unstable CTG repeat located in the 3’-untranslated region (UTR) of DMPK (Figure 4⇓) (49) . DM1 is one member of a class of cognitive diseases whose basis lies in expansion of trinucleotide repeats (see FMR1 and FMR2 below). In the general populace, DMPK contains from 5–27 CTG repeats, whereas in DM1 thousands of repeats can be present.
The molecular basis of DM1 involves the dysregulation of a whole host of genes. Expansion of CTG repeats in the 3’-UTR of DMPK can have several consequences on cellular function. CTG repeats favor nucleosome assembly and a condensed chromatin structure (50) . Indeed, experimental evidence indicates that in DM1, the CTG repeats in DMPK favor condensed chromatin (51) . Condensed DNA is not accessible for binding by non-nucleosomal proteins such as transcription factors. Thus, normal expression of genes near the vicinity of DMPK, such as the homeodomain protein Six5, is most likely disrupted (52) .
The amounts of mature DMPK transcripts in the cytoplasm are significantly reduced in DM1 (53) . Interestingly, the mutated DMPK transcripts, which possess CUG-repeat expansions, are actually retained within the nucleus such that there is no decrease in total DMPK1 mRNA expression in cells from DM1 patients (54) . Retention of mutated DMPK transcripts can have several adverse effects on RNA processing and translation. CUG-rich DMPK transcripts can effectively titrate out CUG-binding proteins involved in RNA metabolism, inhibiting their normal function (55) . In addition, the presence of excessive numbers of DMPK transcripts can act as antisense RNAs by interfering with the expression of other transcripts that contain CAG repeats (56) . Thus, expanded CTG repeats can directly affect the expression of both neighboring genes in the chromosome through chromatin restructuring, and many other genes by inhibiting of RNA processing and translation. Therefore, the molecular effects of the CTG expansion in DM1 are most likely pleiotropic.
Haploinsufficiency of DMPK likely also impacts several cell-signaling pathways. DMPK is a novel serine-threonine protein kinase, with some similarity to the cAMP-dependent protein kinases (57) . The binding of Rac1, a GTPase associated with the actin cytoskeleton, to DMPK stimulates the kinase activity of DMPK in a GTP-dependent manner (57) . Furthermore, the serine-threonine kinase Raf-1 phosphorylates and activates DMPK (57) . Therefore, DMPK could potentially integrate MAPK signals with other signaling pathways.
Fragile X Mental Retardation
Fragile X syndrome is the most commonly inherited form of mental retardation affecting approximately one in 4,000 males (58) . Male phenotypic characteristics include moderate macrocephaly, long-narrow facial features with unusually large ears, and general muscle hypotonia. Mental retardation severity can range from moderate to severe, with most patients showing IQs of approximately fifty. In females, Fragile X is seen in about one in 7,000 live births; however, only approximately 30% of those show any physical characteristics and all affected females exhibit a milder mental handicap.
The Fragile X mental retardation syndrome is similar to other inherited neurodegenerative disorders such as DM1 in that it is effected by an abnormal expansion of repeated trinucleotide sequences within the gene. Genetic analysis has revealed that the trinucleotide expansion often does not appear all at once, but rather occurs over several generations. Trinucleotide expansion ultimately leads to decreased gene expression through mechanisms that include aberrant methylation of DNA, and formation of restrictive chromatin structure.
Fragile X Mental Retardation 2
Expansion of a CCG repeat at Xq28 in the 5’ untranslated region of the FMR2 exon 1 at the Fragile-X E (FRAXE) site results in the methylation and transcriptional disruption of FMR2 (Figure 4⇓) (59, 60) . Deficits in FMR2 differ considerably from FMR1 in affects on behavior and the mechanism of cognitive disruption. FMR2 is a rare disorder that occurs once in every 50,000–100,000 births. FMR2 is associated with mild mental impairment (IQ of 50–85) and primarily manifests as moderate learning difficulties and delay in acquisition of language. A number of variable physical abnormalities are observed, such as a long, narrow face, mild facial hypoplasia, irregular teeth, thick lips, and nasal abnormalities. A subset of FRAXE patients may exhibit behavioral disorders that closely resemble attention deficit and hyperactivity disorder (ADHD), and autism.
Produced through the disruption in the first exon of the homologous murine FMR2 gene, the FMR2 knockout mouse has revealed many interesting possibilities for a role for FMR2 in development. Temporal regulation of wild-type FMR2 is seen during embryogenesis where FMR2 is highly expressed for a brief time in ganglionic eminences of the ventral telencephalon (61) . Beginning on E10.5, transcription of FMR2 coincides with the differentiation of neuroblasts to various neuronal tissues (61) . Neuronal progenitor cells that migrate from the ventral telencephalic region later develop in the cortices, with a large fraction of the progenitor cells maturing into GABAergic interneurons of the neocortex (61) . Thus, in FMR2−/− mice, inhibitory GABAergic neurotransmission may be disrupted.
FMR2 -deficient mice show several interesting phenotypes relevant to memory formation. FMR2 mice have deficits in associative fear-conditioned learning, specifically hippocampus-dependent contextual fear conditioning (61) . In sharp contrast to the observed deficits in hippocampus-dependent learning, NMDA-dependent and -independent LTP was significantly enhanced, even at saturating levels of high-frequency stimulation in hippocampal area CA1 (61) . These results are illustrative of a model for human mental retardation where hippocampus-dependent learning is inhibited, whereas induction and/or expression of hippocampal LTP was enhanced. It is possible that the prominent role FMR2 plays in the development of GABAergic interneurons may underlie the enhancement of hippocampal LTP and deficit in hippocampus-dependent memory formation.
The true function of FMR2 is unknown; however, evidence indicates that FMR2 is an activator of transcription. FMR2 is a member of a protein family that includes AF4, LAF4, and AF5Q31. The members of this family share high, regional amino-acid similarity (20%-35% total amino acid identity) and localize to the nucleus. Moreover, these proteins, including FMR2, significantly augment transcription in both yeast and HeLa cells (62) . However, the mechanism of transcriptional activation employed by the FMR2-like proteins is still unknown. It is possible that other members of the FMR2 family can compensate for specific deficits in FMR2, which could explain the variability in cognitive deficiencies seen in FRAXE.
Dendritic morphology in cognitive deficits
Early studies of synapse morphology in the hippocampus suggested that induction of synaptic plasticity in vitro and memory formation in vivo are associated with increases in the number of synapses (63, 64) . Synapses in the mammalian CNS are located at dendritic spines, and several recent studies have indicated that dendritic spine morphology is very dynamic. Changes in spine morphology are correlated with changes in synaptic activity, such as occurs during induction of LTP in vitro (65) . Vital to dendritic remodeling is the actin cytoskeleton (66) . In the following sections, we review two diseases of human memory formation that also affect the morphology of dendritic spines.
Fragile X Mental Retardation 1
FMR1 is located at Xq27.3 at a unique site on the X chromosome known as the Fragile X A site (FRAXA) (67) . The polymorphic CGG-repeat responsible for the loss of gene expression occurs at the 5’ UTR of FMR1 (Figure 4⇓). Interestingly, if the repeat number is between ∼50 and 200, normal protein is produced and no dysfunction is seen. However, if the expansion exceeds 200 CGG repeats the disease will become manifest; in some cases, more than 1000 CGG repeats in FMR1 have been observed. The mutation of FMR1 results in hypermethylation of the CGG expansion and of the upstream CpG island in the promoter, leading to transcriptional silencing of FMR1 . The instability of the trinucleotide expansion during embryogenesis often leads to significant mosaicism in nearly 15% of patients with harboring at least 200 CGG repeats.
FMR1 mRNA undergoes extensive alternative splicing and can form up to twenty alternatively sliced proteins; however, only four or five protein isoforms are detectable. Almost all FMR1 protein (FMRP) consists of FMR1 isoform 7, which lacks exon 12. The first solid evidence suggesting a specific role for FMRP in cognition came from studies on brains of three fragile X patients that showed an increase in dendritic spine density in the CNS (68) . The dendritic spines had a long, thin morphology, which closely resembled the immature spines observed in developing neurons. This suggested a role for FMR1 in neuronal maturation or development, but it was not until the investigation of a mouse model for FMR1 that putative mechanisms of FMRP action began to emerge.
In 1994, a FMR1 knockout mouse was developed (69) . Although the animal model differs from the human cause of gene disruption, absence of FMRP results in a phenotype resembling that in FMR1 humans. This includes macroorchidism and deficits in spatial learning and associative fear-conditioned learning (70) . There is an overall increase in the number of dendritic spines in FMR1 mice (Figure 5⇓). Dendritic spines of FMR1 mice are long and thin, mimicking the human condition.
FMRP is a cytoplasmic RNA-binding protein that contains three functional RNA binding motifs [two KH (heterogeneous nuclear RNA-binding protein K homology) domains and an RGG (Arg–Gly–Gly) box] and has been shown to associate with G quartets present in mRNA in vivo (71) . There is some evidence that FMRP may chaperone mRNA from the nucleus to the synapse (Figure 5⇓). Another possible role for FMR1 is in synaptic maturation (Figure 5⇓). The increase of long, thin dendritic processes and a higher density of dendritic spines observed in the brains of Fmr1−/− mice and humans with FMR1 suggest that the lack of mature synapses underlies the observed cognitive deficits. The appearance of immature spines may not be due to a developmental derangement, but rather due to changes in protein expression under the transcriptional control of FMRP. This theory is supported by two lines of evidence: 1) The synaptic immaturity seen in FMR1 knockout mice and in human patients resembles that which can be caused by sensory deprivation and, 2) FMR1 translocates to the areas surrounding synapses following stimulation of metabotropic glutamate receptors. A greater understanding of the pathogenesis of the Fragile X syndrome will unquestionably be attained through the analysis of the function of FMRP.
Williams Syndrome
Williams Syndrome (WS) is a relatively common disorder, occurring once in 20,000 live births (72) . WS patients exhibit an interesting idiosyncratic social behavioral syndrome—they are overly friendly. Behavioral studies of WS patients show that they manifest an abnormal positive bias toward unknown individuals (73) ; they have been described as “never having met a stranger,” treating even people that they have just met with great familiarity. They also exhibit spatio-visual processing deficits, highly acute hearing, and mild-to-severe mental retardation. WS arises from deletions in chromosome 7 that typically include multiple genes. Given the complex behavioral manifestations in WS patients, Williams Syndrome provides an interesting example of genetic contributions to complex social behaviors.
WS is associated with a 1.5 Mb deletion in chromosome 7q11.23 (74) . Several genes have been identified within the WS locus (72) including LIM Kinase 1 (LIMK-1) (75, 76) . LIMK-1 is a target of the Rac and Rho small GTPases, and PKC signals that control cytoskeletal organization (Figure 5⇓). LIMK-1 exerts its effects through phosphorylating and inhibiting Actin Depolymerization Factor (ADF)/cofilin. ADF/cofilin normally binds directly to actin and promotes its depolymerization. Thus, the LIMK-1 pathway is one of the specific mechanisms whereby the Rho system regulates cytoskeletal structure.
LIMK-1 homozygous knockout mice, as expected, have alterations in actin microfilaments due to derangements of actin turnover. They also manifest altered dendritic spine morphology at Schaffer collateral synapses in the hippocampus. Specifically, they have fewer actin microfilaments than the normally actin-dense spines, and the dendritic spines in LIMK-1 knockout mice lack the normal bulbous ending that dendritic spines exhibit. There are no apparent alterations in synaptic number or baseline synaptic function, however. Like FMR2 knockout mice, LIMK-1 knockouts exhibit LTP that saturates at a higher level than that seen in control mice—that is, a higher maximal level of potentiation is achieved with repetitive LTP-inducing stimulation. This alteration in synaptic plasticity is associated with impaired learning as well. Specifically, LIMK-1 knockouts show a decrease in their capacity to “unlearn” the location of a hidden platform in a Morris Water Maze task, once they have learned that a platform is associated with a particular location. This is assessed using a platform reversal variation of the Water Maze, where the hidden platform is moved to a new location after the animal has been repeatedly trained with the platform in one place. The effects on LTP and dendritic spine morphology in the LIMK-1 knockout mouse suggest a possible basis for the cognitive effects in WS. They also point to the growing understanding of the importance of morphological regulation in learning and synaptic plasticity, including, apparently, those processes occurring in the human CNS.
Summary of Diseases Affecting Human Learning and Memory.
Conclusions
A great deal of progress has been made in the last decade in understanding the molecular basis for deficiencies in human memory formation. Much of this progress has been made using animal models where normal gene expression has been disrupted. Interpretation of results from knock-out animal models is difficult, as one cannot rule out the possibility that expression of the altered gene may not be directly involved in memory formation, but rather is involved in development of the relevant neural pathways. Most of the mutations we describe do not lead to gross morphological changes in the CNS in the human or mouse. Mouse studies, where available, indicate that baseline synaptic transmission in many of the mutations is normal as well. These observations are consistent with an ongoing need for newly synthesized gene products in cognitive processing. Additionally, in the case of the ERK–CREB–CBP pathway, there is direct evidence from animal studies that acute inhibition of the signal in adults is sufficient to cause learning deficiencies. These types of considerations suggest a rethinking of our outlook on human learning disorders from the traditional view of them as developmental problems to a new view of them as cognitive deficiencies. This change in outlook has been precipitated by new and ongoing discoveries concerning the basic signal transduction processes underlying learning and memory.
Arguably, the holy grail quest of neuroscience research is unlocking the mystery of human cognition. Initial studies of brain structure proved intimidating; how could one find the needle that is human cognition, when it lay buried in a haystack of 1010 neurons with 1013 connections? Studies of brain lesions elucidated where in the brain memories were formed and stored, but shed little light as to the mechanisms underlying its formation. It was not until the molecular genetics revolution that serious progress was made in beginning to dissect the cellular and molecular basis of human cognition. Perhaps the most striking observation made concerning the molecular basis of human cognitive deficiency is that no cellular process is invulnerable to disruption. Deficits in human cognition arise from cellular defects in receptor-mediated second-messenger signaling pathways through disruptions in chromatin structure (Figure 5⇓). In fact, we now live in a world where observations from knockout mice are utilized to understand the associated condition in “knockout” humans. With all the progress that has been made understanding cognition and the basis for deficits, the next wave of research should begin to shed light on how to alleviate, or possibly cure some of these debilitating diseases.
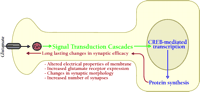
Processes involved in memory formation. Formation of memory is a complex process that requires several aspects of cell function. Memory formation begins at the plasma membrane. Upon activation, NMDA-type glutamate receptors allow Ca2+ to flow into the cell. The Ca2+ flux activates several second-messenger signaling cascades. Ultimately, these signaling processes activate the transcription factor CREB, which leads to the modulation of several genes required for consolidation of long-term memory. Translation of newly synthesized mRNAs leads to production of proteins required to affect long-term changes in neuronal physiology.
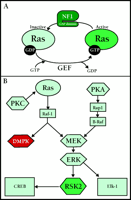
Signaling pathways implicated in human memory formation. A. Ras is a small GTPase whose activity is regulated by the presence of GTP or GDP. When bound to GDP, Ras is inactive. Guanine nucleotide exchange factors (GEF) catalyze the exchange of a molecule of GDP for a molecule of GTP. Once bound to GTP, Ras is activated. GTPase activating proteins (GAP) promote the hydrolysis of GTP to GDP, inactivating Ras. NF1 contains a GAP, and is involved in inactivation of Ras. B. Signaling through PKA and PKC leads to activation of ERK/MAPK. In parallel, Raf-1 also activates the DM1-associated protein kinase (DMPK) involved in myotonic dystrophy. RSK2 is activated by ERK, and regulates gene transcription. CLS is due to disruption of RSK2.
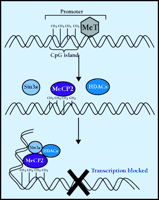
Repression of transcription via methylation. DNA methyltransferases (MeT) catalyze the methylation of cytosine, producing 5’-methylcytosine (5 mC). Most cytosine methylations occur in cytosine phosphorylated guanine (CpG) dinucleotide repeats (islands). The transcriptional repressor, methyl-CpG binding protein 2 (MeCP2), binds to 5mCpG and recruits histone deacetylases (HDAC) and corepressors, such as Sin3a. The activity of this protein complex causes a change in chromatin structure such that transcription is blocked. Rett syndrome is believed to be caused by disruption of MeCP2.
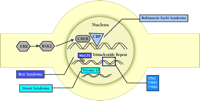
Nuclear function in cognitive disorder. Consolidation of long-term memory requires the regulation of gene expression. Active Rsk2 phosphorylates the transcription factor, CREB. P-CREB recruits the coactivator CREB binding protein (CBP) and promotes transcription of genes whose promoters contain CRE sites. MeCP2 permanently represses expression of some genes that are methylated in CpG islands found in the promoters. Loss of MeCP2 causes an aberrant increase in the dosage of some genes, usually genes that undergo imprinting. Expansion of trinucleotide repeat sequences in genes changes local chromatin structure and influences RNA metabolism, leading to the dysregulation of several genes. Presence of an extra chromsome (trisomy) results in pathological over-expression of potentially hundreds of genes.
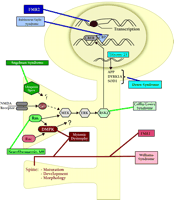
Disruptions along the molecular cascade for memory formation lead to cognitive disorder. Memory formation begins with activation of signaling pathways at the membrane of dendritic spines. The signaling cascade reaches the nucleus, where the activity of transcription factors is modulated, resulting in changes in gene expression. Newly synthesized proteins cause long-term changes in cell function, such as the growth and maturation of new dendritic spines.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

J. David Sweatt, PhD , is a Professor in the Division of Neuroscience and a member of the Mental Retardation Research Center at Baylor College of Medicine. His research focuses on basic mechanisms of learning and memory, mental retardation syndromes and Alzheimer’s disease. Edwin J. Weeber, PhD , is an Instructor at Baylor College of Medicine studying signal transduction mechanisms involved in learning and memory. Jonathan M. Levenson, PhD , is a Post-doctoral Fellow in Dr. Sweatt’s laboratory at Baylor College of Medicine studying the molecular genetics of long-term memory formation in the hippocampus. Address correspondence to JDS. E-mail jsweatt{at}bcm.tmc.edu

