Cytokines in Heart Failure: Potential Interactions with Angiotensin II and Leptin
The hypothesis that the pathophysiology of heart failure involves, in part, an inflammatory process has been tested over the past decade (1,2) . Primarily, these studies have investigated the presence of proinflammatory cytokines and their receptors in the circulation or in cardiac tissue of patients with various etiologies and severity of heart failure. The most consistent finding is that plasma concentrations of tumor necrosis factor–α (TNF-α ) and its circulating receptors are elevated and correlate with the severity of heart failure, as well as with poor prognosis of the patient (2,3) .
Cytokine expression in the myocardium is perhaps more compelling evidence of a proinflammatory process occurring during the progressive remodeling of heart failure. Evidence for the role of proinflammatory cytokines in the process of pathological left-ventricular remodeling comes from studies using transgenic models, as well as from recent research with implanted left-ventricular-assist devices (LVAD) in humans. Transgenic animals that overexpress TNF-α specifically in the myocardium develop fibrotic and dilated hearts with decreased function, and show a fiftyfold increase in interleukin-1β (IL-1β ) gene expression compared to that found in wild-type animals (4) . These changes can be alleviated by infusing the transgenic mice with a TNF-α -specific monoclonal antibody, or by early (prior to onset of cardiac hypertrophy) treatment with batimastat, a matrix metalloproteinase inhibitor (5) . Batimastat is thought to relieve the disease process by decreasing TNF-α -induced matrix protein synthesis, denaturation, and degradation, which contributes to left ventricular remodeling and dilation.
The remodeling of the ventricular wall to a more dilated state marks the progression to end-stage heart failure for the patient and, at the present time, does not appear to be reversible by conventional pharmacological therapies. Recent advances using LVAD provide relief to patients with rapidly decompensating heart failure and allow the left ventricle to begin the process of reverse remodeling. Two dramatic studies have supported the role of proinflammatory cytokines in this pathologic remodeling process and demonstrate the effect of LVAD in decreasing cardiac proinflammatory cytokine levels. Torre-Amione et al. observed that TNF-α is diffusely present in the left ventricle of patients at the time of LVAD implantation, but that the amount of TNF-α decreases nearly 95% in the left ventricle following the use of LVAD for 16–62 weeks (6) . Birks et al. compared the production of TNF-α , IL-1β , and IL-6 in left ventricular samples from patients with decompensating heart failure who required LVAD support to those from patients with less severe heart failure undergoing elective heart transplantation (7) . RNA transcripts of TNF-α and IL-6 were increased twofold and 2.5-fold, respectively, whereas the expression of IL-1β transcripts was increased tenfold in the LVAD patients compared to those observed in patients with New York Heart Association-(NYHA)-defined class III or IV heart failure. The stimulus for the increased production and activity of cytokines is unknown, but it does not appear to be due to increased inflammatory cell activation (8) .
In addition to pressor and hypertrophic effects, recent findings suggest that the neurohormone angiotensin II (AII) may interact with or promote the expression of cytokines to effect the progression of heart failure (9) . The angiotensin converting enzyme (ACE) inhibitor drug class is an essential tool in the treatment of chronic congestive heart failure. Patients treated with ACE inhibitors or AII type I receptor (AT1R) antagonists have decreased concentrations of circulating TNF-α and some studies suggest that higher doses of ACE inhibitors will routinely decrease circulating levels of the proinflammatory cytokine IL-6 (10–13) .
However, other enzymes in addition to ACE are capable of generating AII in the heart, and chymase, which is not blocked by ACE inhibitors, may allow for local generation of AII, decreasing ACE inhibitor efficacy (9) . The intracellular pathways by which AII stimulates cytokine production involve protein kinase C (PKC) and, subsequently, the activation of the transcription factors nuclear factor-κ B (NF-κ B) and activator protein-1 (AP-1) (14) . It is interesting to note that TNF-α and IL-1β are potent inducers of AII receptor expression (15,16) , perhaps setting the stage for a vicious cycle of AII-induced myocardial cytokine production and cytokine-induced increases in AII activity (Figure 1⇓).
The initial discovery that TNF-α levels are elevated in heart failure was not driven by the notion of heart failure as an inflammatory process, but rather, that heart failure patients frequently present with a syndrome known as cardiac cachexia (chronic wasting of adipose tissue and skeletal muscle, while sparing the cardiac muscle). TNF-α (also termed cachectin) was hypothesized to mediate this syndrome (17) , and indeed, the concentration of TNF-α is typically higher in the circulation of cachectic versus non-cachectic heart failure patients (18) . TNF-α is thought to mediate muscle and adipose wasting by activating proteasomes and NF-κ B–dependent pathways in skeletal muscle and adipocytes (19) . The decrease in cardiac function (cardiac suppression) elicited by TNF-α is thought to be mediated through the activation of cytokine-induced nitric oxide synthase (iNOS), ceramidase and sphingomyelin pathways, NF-κ B activation, and the eventual uncoupling of β1 -adrenergic signaling. Interestingly, IL-1β shares the cardiosuppressive pathways of iNOS and ceramidase activation. In addition, IL-1β may also suppress cardiac function by increasing cyclooxygenase-2 (COX-2) and phospholipase A2 gene expression, and by phosphatidylinositol-3’ kinase activation, which results in NF-κ B activation (20,21) .
Subsequent to the discovery by Levine and colleagues of the role of TNF-α in cardiac cachexia (17) , another cytokine, leptin, has been characterized as a major regulator of body mass and appetite (22) . Leptin is primarily produced by adipocytes, whereas its receptors are expressed in a variety of tissues, including the heart (23) . The long form of the leptin receptor is similar to gp130, a member of the IL-6 receptor (IL-6R) family, which also includes the receptors for leukemia inhibitory factor (LIF), and cardiotrophin I (24) . The long form of the leptin receptor is unusual because it forms homodimers exclusively, whereas other members of the IL-6R family are capable of forming homodimers or heterodimers with other members of the gp130 class. Leptin receptors signal primarily by activating Janus kinase 2 (JAK2) and signal transducers and activators of transcription 3 (STAT3) in the majority of tissues studied, and also appear to activate suppressor of cytokine signaling 3 (SOCS3) (25) . The potential for leptin to modulate the activity of other cytokines has been reported (26,27) and this modulation may include interference with NF-κ B effects by STAT3 (19) . The ability of leptin to modulate NF-κ B activity through the phosphorylation of STAT isoforms may also affect the ability of AII to induce production of TNF-α and IL-1β , but this hypothesis has not been tested.
Leptin suppresses cardiac contractility through a nitric-oxide-(NO)-dependent pathway, and this cardiac suppression appears to be blunted in hypertensive animals (28,29) . However, these studies should be interpreted cautiously because the concentrations of leptin that achieved these effects were beyond that which is physiologically attainable. Nonetheless, physiologically relevant concentrations of leptin (25 ng/ml) are capable of completely abolishing IL-1β –induced decreases in cardiac contractility, suggesting a potential role of leptin in modulating proinflammatory cytokine effects in the heart during the progression of heart failure (30) . Recent studies have examined circulating levels of leptin in various stages of heart failure and have compared concentrations in cachectic vs. non-cachectic patients (31–33) . In general, these studies have found that leptin is increased in non-cachectic heart failure patients, but that cachectic patients have normal-to-low circulating levels of leptin. These findings are intriguing if, indeed, leptin is protective against the cardiosuppressive effects of TNF-α or IL-1. By facilitating the decrease of circulating concentrations of leptin, cardiac cachexia may facilitate the downward spiral of cardiac decompensation effected by elevated amounts of TNF-α or IL-1.
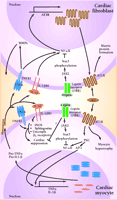
Angiotensin II (AII), acting through angiotensin II type I receptors (AT1R), promotes cardiac myocyte hypertrophy and cardiac fibroblast matrix formation. Activation of AT1R signals the nucleus via protein kinase C (PKC), nuclear factor κB (NF-κB), and activator protein 1-(AP-1)-dependent pathways to increase transcription of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β). Following protein processing, mature TNF-α and IL-1β are secreted and can act in an autocrine or paracrine fashion to activate their respective receptors. In the cardiac fibroblast, TNFR1 and IL1βR1 activation results in increased activity of matrix metalloproteinases (MMP). Additionally, activation of NF-κB can lead to increased AT1R production. In cardiac myocytes, these cytokines bring about a multifaceted suppression of cardiac function by activating nitric oxide synthase (iNOS), increasing sphingosine production, and uncoupling β1-adrenergic receptors. Leptin, by binding to its receptor (OBR), can activate Janus kinase 2 (JAK2). This results in phosphorylation of signal transducer and activator of transcription (STAT3), which can alter the ability of NF-κB to activate gene transcription. These, as well as other possible leptin-induced processes, have the potential to disrupt AII-cytokine pathways, thus modulating cytokine-induced cardiac suppression.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

M. Judith Radin, DVM, PhD , is an Associate Professor in the Department of Veterinary Biosciences and an Associate Investigator in the Dorothy M. Davis Heart and Lung Research Institute at The Ohio State University. She is a Veterinary Clinical Pathologist and a diplomate of the American College of Veterinary Pathologists.

Bethany J. Holycross, PhD , is a Research Scientist in the Department of Molecular and Cellular Biochemistry and the Dorothy M. Davis Heart and Lung Research Institute at The Ohio State University. Address correspondence to BJH. Email gailliot.4{at}osu.edu.

