Rapid Extranuclear Signaling by the Estrogen Receptor (ER): MNAR Couples ER and Src to the MAP Kinase Signaling Pathway
The estrogen receptor (ER), a member of the superfamily of ligand-dependent transcriptional activators, consists of a C-terminal steroid ligand–binding domain (LBD), a centrally located DNA binding domain (DBD), and an N-terminal domain of less well-characterized function. Additionally, the ER contains two autonomous transcriptional activation domains (AF): the AF-1 domain, located in the N terminus, and AF-2, located within the LBD. Steroid receptors are latent transcriptional activators that require ligand binding for activation. Steroid binding induces a specific conformational change(s) that results in receptor dissociation from a protein chaperone complex, dimerization of steroid receptors, and binding of the receptor dimer to specific steroid response elements (SREs) located in the 5′ regulatory region of primary steroid-responsive target genes. Through interaction with AF-1 or AF-2, the activated, DNA-bound receptor recruits transcriptional coactivators that mediate assembly of a productive transcription complex at the promoter. Thus, the major actions of estrogen have long been held to be the result of direct transcriptional regulation of specific gene networks (1).
Not all actions of estrogen can be explained by the ER’s direct transcriptional regulation of target gene networks. Estrogens, and for that matter all classes of steroid hormones, induce rapid activation (seconds to minutes) of various signaling molecules and signal transduction pathways independent of synthesis of new mRNA and protein. This rapid action of steroids in the absence of gene transcription and protein synthesis is termed “nongenomic” or “nontranscriptional” signaling (2–4). The specific signaling response induced by estrogen is dependent on the biological status of specific tissues and cells. For example, estrogen rapidly activates the Shc-Src-Ras–Raf-MAP [mitogen-activated protein kinase; also known as extracellular signal-related kinases (Erk)1 and Erk2] phosphorylation signaling cascade in breast cancer cells, which may elicit the proliferative response (5–7). The “proliferative” effect of estrogen in breast cancer cells also involves rapid estrogen-induced activation, in a Src-dependent manner, of the heterodimeric PI3K–Akt antiapoptotic signaling pathway (8) (i.e., the phosphoinositide 3-kinase–protein kinase B antiapoptotic pathway).
The receptor mechanism that mediates rapid nongenomic actions of estrogen is a controversial issue and has not been well defined. The controversy entails whether a separate membrane receptor for estrogen exists that is unrelated to the classical ER, or whether a subpopulation of the classical ER associates with the cell membrane to mediate rapid nongenomic actions of estrogen. A membrane receptor for estrogen unrelated to ER has neither been isolated nor characterized. Estrogen has been reported to rapidly activate MAPK in certain ER-negative breast cancer cell lines in a manner dependent on the orphan G protein–coupled receptor GPR30 (9). Nonetheless, whether GPR30 directly binds estrogen or functions as a bona fide receptor has not been determined.
There is growing evidence for the involvement of a subpopulation of the classical ER in the mediation of estrogen’s nongenomic effects: specific ER antagonists are effective inhibitors of most rapid estrogen responses (5–8, 10–12) ; reconstitution of rapid estrogen signaling in most ER-negative cell lines requires ectopic expression of ER; a fraction of the total endogenous ER pool rapidly translocates to areas of membrane ruffles in response to short-term estrogen treatment of MCF-7 breast cancer cells (7) ; endogenous ER has been isolated biochemically in purified plasma membranes and caveolae of endothelial cells, and rapid responses to estrogen in isolated membranes are dependent on the presence of ER (13) ; and finally, rapid estrogen signaling responses appear to be due to an extranuclear action of ER, based on experiments using cell compartment–specific tagged ER (10, 14). ER targeted to cell nuclei fails to mediate the antiapoptotic effects of estrogen, whereas ER targeted to the cell membrane exhibits the same ability to mediate rapid effects of estrogen as wild-type ER. The results of these cell compartment–targeting experiments also reveal that the transcriptional and nontranscriptional actions of ER are separable: membrane-targeted ER cannot mediate estrogen-dependent regulation of SRE-containing genes, whereas nucleus-targeted ER can do so.
Analyses of various mutant forms of ER have also revealed that transcriptional and nontranscriptional functions are separable in the same ER molecule. The core LBD, which is inactive as a transcription factor, is sufficient for mediating many of the rapid estrogen responses in signaling pathways (8, 9, 13, 15, 16). The process by which a portion of the classical ER localizes to the cell membrane remains unresolved. Steroid receptors rapidly shuttle between the nucleus and cytoplasm by active nuclear import and export mechanisms. In the absence of identified posttranslational modifications that could be involved in attachment of ER to the cell membrane, it is likely that membrane translocation of ER is facilitated by another protein (4) such as caveolin-1, which has been reported to interact with ER (14). These data collectively support the concept that ER is a dual function protein capable of acting as a transcription factor or signaling molecule that mediates rapid actions of estrogen in signal transduction pathways. Which hat ER wears likely depends on its intracellular localization (Figure 1⇓).
Important unresolved questions are how ER interacts with signaling molecules in an estrogen-dependent manner and how this interaction can trigger a signaling response. ER itself does not have protein kinase activity, and specific motifs for interaction with regulatory domains of signaling molecules have not been identified. Although an estrogen-dependent interaction of ER with Shc, Src, and PI3K has been reported, it is unknown whether this interaction is sufficient to stimulate kinase activities in vivo or whether other factors are required. In vitro, ER is able to interact with the SH2 domain of Src (8, 17, 18), but this does not appear to be sufficient for activation of Src (18, 19). A recent paper by Wong et al. describes the identification of modulator of nongenomic activity of estrogen receptor (MNAR) as an adaptor protein between ER and Src and provides a potentially important breakthrough in explaining the molecular mechanism of how ER interacts with and activates Src (19). MNAR, identified during the course of a biochemical-proteomic screen for ER-interacting proteins, is a 120-kDa protein that is most closely related to PELP1 (proline-, glutamic acid–, leucine rich–protein). PELP1 was initially identified as a specific phosphotyrosine-independent ligand for the Src homology domain 2 (SH2) domain of the protein tyrosine kinase lck and was later shown to act as coactivator for ER (15). MNAR facilitates and stabilizes the estrogen-dependent ER–Src interaction by forming a ternary complex with ER and Src, and enhances estrogen- and ER-dependent stimulation of Src activity both in vitro and in transfected mammalian cells. MNAR contains LXXLL and PXXP motifs, suggesting a mechanism for how MNAR can function as an adaptor between ER and Src. The AF-2 domain of ER interacts, in a hormone-dependent manner, with LXXLL motifs contained in a family of 160-kDa (p160) steroid receptor coactivators. Estrogen agonists induce a distinct conformational change in the most C-terminal α helix of AF-2 that creates a hydrophobic pocket for interaction with LXXLL motifs (1). Interestingly, estrogen antagonists induce an alternate conformation in AF-2 that does not permit this interaction (1), suggesting a mechanism for how ER antagonists block rapid estrogen responses on Src initiated signaling pathways. Src is autoregulated by intramolecular interactions between the SH2 domain and a pTyr537 in the C-terminal tail, and between the SH3 domain and PXXP-like motifs in the linker region that connects the regulatory domain with the catalytic lobes (Figure 2⇓). These intramolecular interactions maintain Src in a closed inactive conformation. Conversion to the open, kinase-active conformation occurs by competition with external peptide ligands, causing displacement of the internal SH2 or SH3 domain interactions (16). Figure 2⇓ proposes that MNAR facilitates estrogen- and ER-dependent activation of Src by providing a PXXP sequence that displaces the intramolecular occupation of SH3 in Src. Interaction of ER with the SH2 domain of Src may provide additional stabilizing interactions to contribute to Src activation (Figure 2⇓).
In support of this model, MNAR potentiates the direct transcriptional activity of ER, but this effect is blocked by inhibitors of Src and MAP kinases. These inhibitors do not affect the activity of p160 coactivators; thus, it is not likely that MNAR functions as a conventional ER coactivator. Potentiation of the transcriptional activity of ER is likely due to positive feedback between the estrogen-stimulated Src-Erk signaling pathway and the ER transcription complex (Figure 1⇓). Previous studies have shown that activation of MAPK signaling pathways can stimulate the transcriptional activity of ER by phosphorylation and activation of AF-1 (20).
The identification of MNAR has created exciting opportunities for further study and provides potentially important new insights into the crosstalk between classical ER and signal transduction pathways. However, experiments are needed to prove the proposed mechanism of how MNAR physically and functionally couples ER with Src shown in Figure 2⇓. Additional experiments are also needed to determine the role of MNAR in mediating cell biological responses to estrogen such as cell proliferation, apoptosis, and survival. The paper by Wong et al. only examines the influence of MNAR on the Src-Erk signaling pathway (19). How MNAR expression is regulated also needs to be determined. Could alterations in relative amounts of MNAR in breast tumors play a role in the sensitivity of ER to synthetic ligands used for treatment or prevention of breast cancer? It will also be interesting to determine the role of MNAR in mediating rapid signaling actions of other steroid hormones. The progesterone receptor (PR) contains a PXXP motif in its N-terminal domain that directly interacts with SH3 domains of Src to induce its progesterone-dependent activation (18). PR is the only steroid receptor that contains a PXXP motif, suggesting that it may not require an adaptor such as MNAR and may have evolved a more direct mechanism for activation of Src. It will also be important to determine whether MNAR and p160 coactivators compete for interaction with AF-2 through LXXLL motifs and if so, how this competition might affect the relative balance between transcriptional and nontranscriptional activities of ER. Could MNAR also play a role in facilitating translocation of a subpopulation of ER to the cell membrane? The intracellular localization of MNAR and how it may traffic in the cell with ER also needs to be determined.
Most of the studies to date on nongenomic actions of ER have been done in vitro with cell culture systems. A few experiments have now been reported suggesting that the rapid nongenomic signaling actions of ER have a role in nonreproductive target tissues in vivo. Vascular protection by estrogen in ischemia-reperfusion injury requires ER-induced activation of eNOS, as mediated by the PI3K-Akt signaling pathway (11, 12). Recently described ER ligands that dissociate the transcription from nontranscription actions of ER have been identified. A synthetic compound termed estren (4-estren-3α,17β -diol) induces only nontranscriptional activities of ER, whereas another pyrazole compound induces the transcriptional activities of ER with minimal effects on its rapid signaling activity. The estren compound administered to mice is equally as effective as estradiol in preventing ovariectomy-induced apoptosis of osteoblasts and in preserving bone density, but estren treatment could not compensate for reduced uterine weight (21). Additional evidence indicates that the expression of subsets of endogenous estrogen-regulated genes are induced through nongenomic ER activation of Src-Erk and PI3K-Akt signaling pathways that converge upon and activate other nuclear transcription factors (9, 22–24). Thus, nongenomic ER signaling might regulate specific gene networks that either complement or broaden the genes directly regulated by ER working as a nuclear transcription factor. As we learn more about the mechanisms and in vivo roles of these two ER pathways in different tissues, it may be possible to develop ER ligands that selectively target the transcription or nontranscription signaling pathways for different therapeutic purposes. It might turn out that MNAR participates in selectively inducing the nontranscriptional activities of ER through distinct ligand-induced conformational changes in the AF-2 region that favor interaction with MNAR over direct transcriptional coactivators.
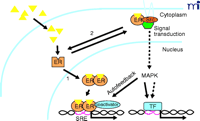
Model of classical and “nongenomic” actions of the estrogen receptor (ER). Estrogenic ligands (triangles) activate ER as a transcription factor (1) by inducing a conformational change(s) that leads to nuclear translocation, dimerization, and binding to specific steroid receptor response elements (SRE) in promoters of primary target genes. Activated ER recruits coactivators that are essential for assembly of a productive transcription complex at the promoter. Alternatively, a subpopulation of ER associates in an estrogen-dependent manner (2) with cytoplasmic- and/or cell membrane–signaling molecules including the tyrosine kinase Src. This extranuclear interaction promotes the Shc-Src-Raf–MAPK kinase (MEK)–MAP kinase phosphorylation cascade. Because MAPK can directly (solid arrows) or indirectly (dashed arrows) activate other transcription factors (TF), the MAP kinase pathway can potentially regulate distinct or complementary sets of genes from those regulated by nuclear ER pathways. Estrogen-activated MAPK can also enhance the direct transcriptional activity of ER by an autofeedback loop through phosphorylation of ER or ER-associated coactivators. The hexagon indicates an adaptor protein.
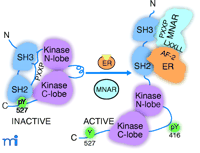
Activation of Src by the ER and the MNAR adaptor protein. ER, in the presence of estrogen (E2), interacts with MNAR and Src to form a stable ternary complex through a specific estrogen-induced interaction between the AF-2 domain in ER and LXXLL motifs in MNAR. Additional stabilizing interactions are provided by the ER interaction with the SH2 domain of Src. PXXP motifs in MNAR interact with the SH3 domain of Src and convert Src from an inactive “closed” conformation to an active “open conformation.” See text for details.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Viroj Boonyaratanakornkit, PhD, is an Instructor in the Department of Pathology, University of Colorado Health Sciences Center, and is a senior member of the Edwards laboratory.

Dean P. Edwards, PhD, is a Professor in the Department of Pathology, University of Colorado Health Sciences Center, Denver, and is Director of the Interdepartmental Program in Molecular Biology and a member of the University of Colorado Cancer Center Program in Hormone Related Malignancies. Address comments to DPE. E-mail Dean.Edwards{at}UCHSC.edu; fax 303-315-6721.

