Portrait of a Killer: The Mitochondrial Apoptosome Emerges From the Shadows
Abstract
Apoptosis (programmed cell death) is a physiological process used to eliminate superfluous, damaged, infected, or aged cells in multicellular organisms. During apoptosis the cellular architecture is dismantled from within in a highly controlled fashion. Members of the caspase family of cysteine proteases are responsible for the destructive phase of apoptosis. One major pathway to caspase activation involves the formation of a multisubunit protease activation complex called the apoptosome. The apoptosome is assembled in response to signals that provoke mitochondrial outer membrane permeabilization and the release of cytochrome c into the cytosol. Recent studies indicate that the apoptosome is a wheel-like structure consisting of seven molecules of Apaf-1 and a similar number of caspase-9 dimers. Knowledge of the structure of the apoptosome will likely lead to the design of therapeutic modulators of apoptosis.

Introduction
Cells destined to die during development undergo an orchestrated program of destruction, a process termed apoptosis. Cells also undergo apoptosis in many other contexts: during the resolution of immune responses, as a result of infection, in response to DNA damage, or as a result of growth factor deprivation. Apoptosis is typically invoked where it is desirable to eliminate cells while minimizing damage to surrounding viable tissue. Accumulating evidence indicates that complex molecular processes control apoptosis and that disruption of these processes may underlie various disease states such as neurodegeneration, autoimmunity, and cancer. Uncontrolled proliferation and reduced sensitivity to apoptotic signals are hallmarks of oncogenic transformation and autoimmune conditions. On the other hand, excessive and inappropriate apoptosis may contribute to neurodegenerative conditions such as Alzheimer disease. Emerging pathways to caspase activation in apoptosis are therefore potential targets for therapeutic intervention.
Caspases: The Executioners of Apoptosis
Apoptosis is characterized by a series of morphological changes, including chromatin condensation, membrane blebbing, and cell shrinkage, which ultimately leads to fragmentation of the cell into small vesicular bodies that are taken up by macrophages (1). Central to the destructive phase of apoptosis is a family of cysteine proteases, called caspases (cysteine aspartic acid–specific proteases), that cleave proteins bearing a conserved amino-acid sequence–motif located N-terminal to a specific aspartic acid residue (Figure 1⇓). Caspases can be broadly divided into two functional subgroups: those that are activated during apoptosis (caspases -2, -3, -6, -7, -8, -9, -10) and those that have been implicated in the processing of proinflammatory cytokines during immune responses (caspases -1, -4, -5). However, the boundaries between these subgroups are not sharply drawn—we still know surprisingly little concerning the roles of several caspases (e.g., caspases -2, -4, -5, -7, -14) in apoptosis or inflammation.
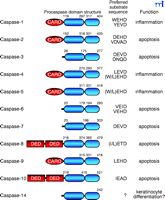
The human caspase family. Domain structures, sites of internal processing, preferred peptide substrate sequences, and biological function of the human caspases are illustrated. Note that each procaspase consists of one large and one small domain and may also include caspase-recruitment domains (CARDs) and death effector domains (DEDs).
Caspases are synthesized as inactive zymogens (procaspases) that require proteolytic cleavage to form the large and small subunits of the active enzyme, suggesting that caspases either become activated through autoproteolysis or by other activated caspases. In fact, both scenarios have been observed.
The fact that procaspases are activated by proteolysis suggests the possibility of a caspase signaling cascade, similar to kinase signaling cascades. Indeed, examples of caspase signaling cascades are now well established. For example, engagement of Fas/APO-1 [a member of the tumor necrosis factor receptor (TNFR) superfamily] by Fas ligand (FasL) induces the recruitment of procaspase-8 to the Fas receptor complex via the adaptor protein FADD (Fas-associated death domain–containing molecule) (Figure 2⇓). Upon recruitment to the receptor complex, caspase-8 becomes activated through autoproteolysis and subsequently cleaves and activates caspase-3, setting up a chain of further caspase activation events (Figure 2⇓). In certain cells, caspase-8 also initiates the mitochondrial (sometimes referred to as “intrinsic”) pathway to caspase activation by cleaving Bid, a member of the Bcl-2 family (Box 1). Cleaved Bid translocates to mitochondria and provokes the release of mitochondrial factors that amplify the death signal through the assembly of a caspase-9–activating complex, called the apoptosome (see below). Assembly of the apoptosome can also be triggered by divergent forms of cellular stress that converge upon mitochondria to permeabilize the outer mitochondrial membrane (1).
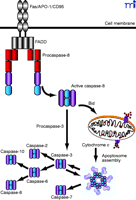
Caspase activation pathways. Initiator caspases are activated through protein–protein interactions, whereas effector caspases are activated through cleavage by initiator caspases. Two major pathways to caspase activation are illustrated. Binding of Fas ligand (FasL) to Fas induces oligomerization of caspase-8 through the adapter molecule FADD. Activated caspase-8 processes and activates caspase-3, leading to further caspase activation events and substrate proteolysis. In certain cells, caspase-8 also activates the mitochodrial pathway by cleaving the Bcl-2 family member Bid, leading to the release of cytochrome c from the intermembrane space and formation of the apoptosome. FADD, Fas-associated death domain–containing molecule. Bid, Bcl-2 homology 3–interacting domain death agonist. Apoptosome reprinted from Acehan et al. Molecular Cell 9, 423–432 (2002), with permission from Elsevier.
Intrinsic versus extrinsic pathways of apoptosis
Activation of intiator caspases during apoptosis can be provoked by extrinsic or intrinsic stimuli. Caspase activation through the extrinsic pathway involves the binding of extracellular ligands [such as FasL or tumor necrosis factor–α (TNFα)] to their respective transmembrane death-evoking receptors. Engagement of these receptors with their ligands provokes the recruitment of the adaptor molecule FADD (Fas-associated death domain) that, in turn, recruits and promotes the activation of caspase-8. Active caspase-8 then proteolytically cleaves (and thereby activates) caspase-3, provoking further caspase activation events that culminate in substrate proteolysis and cell death. Alternatively, stimuli that cause cell stress or damage (such as cytotoxic drugs, heat shock, or radiation) commonly provoke the release of cytochrome c from the mitochondrial intermembrane space into the cytoplasm. In the cytoplasm, cytochrome c can trigger the intrinsic pathway to apoptosis by provoking the assembly of a large caspase-9–activating complex called the apoptosome. Active caspase-9 propagates a proteolytic cascade, ultimately leading to cell death. In some situations, extrinsic death signals can crosstalk with the intrinsic pathway through caspase-8–mediated proteolysis of a small molecule called Bid (a protein that associates with the antiapoptotic protein Bcl-2). Enzymatically processed Bid (called tBid) can provoke mitochondrial cytochrome c release and promote apoptosome assembly.
The Killer from Within: The Mitochondrial Pathway to Apoptosis
In addition to its role as an energy-generating organelle, the mitochondrion has recently emerged as a convergence center for life and death signals (1–,5). A key event in the mitochondrial pathway to apoptosis is the coordinated release of several proteins from the mitochondrial intermembrane space into the cytosol (2–,5). In this pathway, cellular stresses such as DNA damage, heat shock, oxidative stress, and many other forms of cellular damage, result in caspase activation through the release of cytochrome c from mitochondria (5). Cytochrome c exerts its effects through acting as a cofactor for the assembly of a large caspase-activating complex in the cytosol that has been termed the apoptosome (Figure 2⇑).
The discovery of the apoptosome has its roots in seminal work conducted by Wang and colleagues using a cell-free system based on cytosolic extracts from HeLa cells (5). Having first identified a role for mitochondrial cytochrome c in activating caspases, Wang and colleagues then purified the factors that assisted cytochrome c in its deadly mission. Their search led them to Apaf-1, the first identified mammalian homolog of CED-4, a protein known to participate in programmed cell death in C. elegans (6).
Apaf-1 consists of three functional domains: an N-terminal CARD (caspase-recruitment domain), a central nucleotide-binding domain similar to that found in the C. elegans protein CED-4, and twelve to thirteen WD-40 repeats at the C terminus of the molecule (Figure 3⇓). Apaf-1 recruits and activates procaspase-9 through the interactions of CARDs found on both proteins. The precise composition of the apoptosome—the stoichiometry of its components and their spatial symmetry—is still far from clear; however, recent studies have shed some light upon these issues (7).

Domain structure of Apaf-1. The caspase recruitment domain (CARD), nucleotide-binding domain (NBD), and WD-40 repeat regions are indicated.
The Three-Dimensional Anatomy of the Apoptosome
Using electron cryomicroscopy, combined with a variety of previously published data including the crystal structure of the Apaf-1–caspase-9 CARD:CARD complex, Akey and colleagues found that the apoptosome complex is a wheel-like structure comprising a central hub, connected to seven radial spokes (Figure 4⇓) (7, ,8). A model of the apoptosome at 27 Å resolution predicts that the central hub domain is composed of seven Apaf-1 CARD domains held together in close proximity, presumably by an adjacent region previously identified as the Apaf-1 oligomerization surface (7, 9, ,10). In addition, this CED-4–homologous region not only promotes oligomerization, but also forms spokes projecting outward from the hub, connecting the core of the apoptosome to the Y-shaped tail formed by a bundle of WD-40 repeats (Figure 4⇓) (7). Based on the propeller structures of other WD-40 repeat-containing molecules, such as Transducin Gβ, Acehan and colleagues predict that the WD-40 repeats of Apaf-1 form a Y-like tail extending from the bent elbow of the arm formed by the CED-4–homologous region of Apaf-1 (Figure 4⇓) (7, 11, ,12). Molecular modeling experiments suggest that within this Y-shaped tail, the Apaf-1 WD-40 repeat domain is composed of two lobes, the larger of which forms a seven-bladed β -propeller, with the other lobe comprising a smaller six-bladed propeller (7).
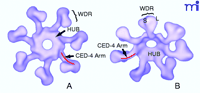
Three-dimensional model illustrating top (A) or oblique (B) views of the apoptosome structure. The central hub domain, consisting of the Apaf-1 CARD and CED-4–homologous regions is indicated, as is the WD-40 repeat (WDR) region that forms a Y-shaped tail. The small (S) and large (L) lobes of the WD-40 repeats are indicated. Note that the CED-4–homologous “arm” region (indicated in red) forms part of the hub domain as well as connecting the hub to the lobes of the WD-40 repeats. Reprinted from Acehan et al. Molecular Cell 9, 423–432 (2002), with permission from Elsevier.
Although Akey and colleagues initially utilized apoptosome complexes assembled in the absence of caspase-9, subsequent in vitro experiments performed in the presence of a noncleavable caspase-9 mutant reveal a similar wheel-like assembly, where caspase-9 occupies a dome-like position sealing the top of the apoptosome hub (7). With procaspase-9 incorporated within the complex, the central hole of the hub formed by the Apaf-1 CARDs is widened slightly. Crudely put, the apoptosome is not too dissimilar to a seven-legged spider with upturned feet, where caspase-9 forms the head of the spider sitting atop a body consisting of seven Apaf-1 molecules arranged in a ring. The Apaf-1 nucleotide-binding and oligomerization domains contained within the CED-4–homologous region likely hold the structure together, whereas the C-terminal WD-40 repeats extend away from the hub.
The crystal structures of several caspases have been solved, including caspase-1, -3, -7, and -8 (13). In the active conformation, all of these caspases appear to exist as heterotetramers, formed through association of two caspase heterodimers. Each caspase heterodimer consists of a large and a small caspase subunit, both of which contribute residues to form the active site. Intriguingly, as suggested by crystal structure analysis, only one of the active sites of a functional caspase-9 dimer may retain proteolytic activity (14). Akey and colleagues speculate that upon apoptosome assembly, the catalytic sites of as yet inactive caspase-9 monomers extend into solution (7). Indeed, the fact that a large part of caspase-9 is not visible in the 3D map of the apoptosome suggests that Apaf-1-complexed caspase-9 molecules might be relatively mobile (7). The increased local concentration of Apaf-1–bound caspase-9 molecules may then facilitate the recruitment of caspase-9 monomers from solution. As a result, the apoptosome-bound caspase-9 molecules may retain catalytic activity, whereas their non-Apaf-1–complexed dimeric partners remain catalytically inactive (7).
Active versus Inactive Apoptosomes
Previous attempts to ascertain the composition and size of the apoptosome have relied largely upon gel filtration analysis (15–,17). The latter studies indicate that apoptosomes, assembled either in vitro, or detected in dying cells, are found in two forms: ∼700-kDa apoptosomes that are proteolytically active (i.e., are capable of activating caspases-3 and -7) or an apparently inactive ~1.4-MDa apoptosome (15–,17). It is currently unclear why the 1.4-MDa form of the apoptosome is functionally inactive (15–,17). One possible clue provided by electron cryomicroscopy studies is that apoptosomes assembled in the presence of caspase-9 appear to be more prone to aggregation (7). It is conceivable that the inactive 1.4-MDa complex is composed of two apoptosomes associated “head-to-head,” with interactions between constituent caspase-9 molecules of individual apoptosomes occuring between the two Apaf-1 layers. Thus, the lack of activity associated with the 1.4-MDa apoptosome may result from hindering the access of potential caspase-9 substrates (such as caspase-3) to the apoptosomal proteolytic core. Indeed, such structures have been observed when apoptosomes were formed in the presence of catalytically inactive procaspase-9 (7). Interestingly, the latter apoptosomes were approximately 1.4-MDa and capable of cleaving procaspase-3, although the activity of this complex in comparison to the more abundant ~1-MDa apoptosomes was not determined (7).
The Role of CytochromecanddATP in Apoptosome Assembly
It is well established that the C-terminal region of Apaf-1, which contains the WD-40 repeat motifs, performs an important regulatory role by repressing the oligomerization of Apaf-1 and the recruitment of caspase-9 to the Apaf-1 CARD motif (1, 9, ,10). On the basis of the low-resolution apoptosome model, the forked structure formed by the two β -propellers of the Apaf-1 WD-40 repeat region could provide a space suitable to accommodate a single cytochrome c molecule (7). It has also been proposed that the CARD region of Apaf-1 is of a similar size to cytochrome c, suggesting that the CARD of autoinhibited Apaf-1 might be buried between the lobes of the WD-40 repeats (7). Although these proposals remain highly speculative, they are broadly supported by a number of observations. First, rotary shadowing experiments suggest that nonactive Apaf-1 monomers may adopt a compact shape (7). In addition, a study by Nunez and colleagues suggests that the Apaf-1 CARD region can associate with the WD-40 repeats, providing a basis for the compact shape of autoinhibited Apaf-1 monomers (9). Also, the observation of Wang and colleagues that binding of procaspase-9 to the Apaf-1 CARD enhances apoptosome assembly supports the concept that cytochrome c –mediated displacement of the Apaf-1 CARD, coupled with caspase-9-binding, prevents Apaf-1 from reassuming the compact, autoinhibited monomeric state (Figure 5⇓) (7, ,18). Interestingly, Akey and colleagues propose that the nucleotide-binding region containing the Walker boxes (a conserved amino-acid motif responsible for ATP/dATP binding) may potentially interact directly with the Apaf-1 CARD, as well as with the CED-4-homologous region of an adjacent monomer (7). Thus, the binding of dATP—already known to be required for Apaf-1 oligomerization (18) —may lock Apaf-1 molecules in an open configuration that facilitates apoptosome assembly (Figure 5⇓).
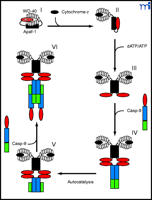
Hypothetical model illustrating the role that cytochrome c and dATP binding play in regulating apoptosome assembly. In the absence of cytosolic cytochrome c, Apaf-1 may exist in a compact, autoinhibited form, with the Apaf-1 CARD region buried between the lobes of the WD-40 repeats (I). The binding of cytosolic cytochrome c then displaces the Apaf-1 CARD from the WD-40 repeats, forcing the molecule into a more open conformation (II). Binding of dATP to the Walker boxes within the CED-4 arm region creates a less flexible, locked conformation (III). This prevents reassociation between the hub and WD-40 repeats, facilitating apoptosome assembly. Caspase-9 (Casp-9) is recruited to the apoptosome via CARD-CARD interaction (IV). Binding to Apaf-1 results in an increase in caspase-9 activity, autoprocessing at D315, and separation of the large and small subunits (V). A free caspase-9 molecule is recruited to each of the Apaf-1–bound caspase-9 molecules; however, only one caspase-9 molecule within each caspase-9 heterotetramer may be catalytically active (VI).
The Apoptosome-Initiated Caspase Activation Cascade
Upon activation of caspase-9 within the apoptosome, the death signal is propagated through the stepwise activation of additional downstream caspases (19–,21), whereby caspases-3 and -7 appear to be simultaneously activated by caspase-9 (19). Indeed, caspase-3 appears to be recruited and retained within the apoptosome by caspase-9 (22). Upon activation by the apoptosome, caspase-3 then processes and activates caspases-2 and -6, which is rapidly followed by the activation of caspases-8 and -10 (by caspase-6). Furthermore, a positive feedback loop between caspases-3 and -9 exists such that activation of caspase-3 rapidly leads to a somewhat differently processed form of active caspase-9 (Figure 2⇑). Thus, upon assembly of apoptosome, rapid amplification of the death signal then occurs through activation of a panoply of other caspases that proceed to dismantle the cell.
Foiling the Death Signal: Negative Regulation of the Apoptosome
A number of controls exist to delay or alter apoptosome-dependent cell death pathways. Members of the inhibitor of apoptosis (IAP) family of proteins, such as XIAP, cIAP-1, and cIAP-2, suppress apoptosis through the direct inhibition of caspases (23). Overexpression of XIAP, cIAP-1, or cIAP-2 in cells interferes with the normal activation of caspases -3, -7, and -9 that occurs downstream of apoptosome assembly (23). Heat shock proteins, such as hsp70 and hsp27, have also been implicated in the suppression of apoptosis downstream of apoptosome assembly. Hsp70 has been reported to prevent caspase activation within the apoptosome by directly associating with Apaf-1, thereby blocking procaspase-9 recruitment (24, ,25).
The most effective inhibitors of the apoptosome pathway might belong to the Bcl-2 family (26). Bcl-2 and its close relatives, such as Bcl-xl, block apoptosis by antagonizing the release of cytochrome c (27, ,28) and other mitochondrial constituents (29), thereby preventing assembly of the apoptosome. However, recent studies suggest that Bcl-2 may also exert its protective effects independent of its ability to antagonize apoptosome assembly (30).
One common strategy for the design of small-molecule enzyme inhibitors involves the molecular modeling of substrate-binding pockets. Indeed, many of the effector caspases were cocrystallized with inhibitory peptides, and these peptides have been used very successfully as caspase inhibitors in vitro. However, as an in vivo therapeutic strategy, inhibition of apoptosome formation may be much more desirable than strategies aimed at neutralizing effector caspases activated downstream. Inhibition of apoptosome formation may be achieved by inhibiting cytochrome c release from mitochondria, or by blocking oligomerization of Apaf-1. The availability of structural data on apoptosome composition is likely to facilitate the design of small molecules to achieve the latter.
Concluding Remarks
It is now clear that the Apaf-1 apoptosome is an important caspase-activating complex in the context of diverse forms of cellular stress. Thus far, studies on the composition of the apoptosome have utilized either recombinant proteins to generate “stripped down” apoptosomes or crudely purified protein fractions isolated from apoptotic cells. Further work on “native” apoptosomes is needed to confirm the composition of the complex and to identify any additional components that may exist.
Acknowledgments
We are indebted to Science Foundation Ireland and the Wellcome Trust for support of ongoing research in our laboratory. SJM is a Science Foundation Ireland Fellow.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Seamus J. Martin, PhD, is a Science Foundation Ireland Fellow and Smurfit Professor of Medical Genetics in the Department of Genetics, Trinity College Dublin, Ireland, and is Director of The Molecular Cell Biology Laboratory within the same department. E-mail: martinsj{at}tcd.ie; fax +353 1 679 8558

Colin Adrain, PhD, received his doctorate from Trinity College Dublin, Ireland in 2001 and is currently a senior postdoctoral fellow in the Martin laboratory, Trinity College, Dublin.

Michelle M. Hill, PhD, received her doctorate from the University of Queensland, Australia in 2000, conducted a postdoctoral fellowship in the laboratory of Dr Brian Hemmings in the Friedrich Miescher Institute, Switzerland and is currently a senior postdoctoral fellow in the Martin laboratory, Trinity College, Dublin.

