Recent Advances in the Neurobiology of Schizophrenia
Abstract
Despite great progress in basic schizophrenia research, the conclusive identification of specific etiological factors or pathogenic processes in the illness has remained elusive. The convergence of modern neuroscientific studies in molecular genetics, molecular neuropathology, neurophysiology, in vivo brain imaging, and psychopharmacology, however, indicates that we may be coming much closer to understanding the molecular basis of schizophrenia. Schizophrenia may be a neurodevelopmental and progressive disorder with multiple biochemical abnormalities involving the dopaminergic, serotonin, glutamate, and γ -aminobutyric acidergic systems. In the near future, biological markers for the illness may come from the combination of diverse assessment techniques. An understanding of the pathophysiology of schizophrenia will be essential to the discovery of preventive measures and therapeutic intervention. Rapidly advancing research into schizophrenia includes diverse etiological hypotheses, and offers directions for future research and treatments.
Introduction
Schizophrenia is a relatively common, chronic, and frequently devastating neuropsychiatric disorder, affecting about one percent of the world’s general population (1). It imposes a disproportionately large economic burden in terms of hospitalization, chronic treatment and rehabilitation, and lost productivity (2). The onset of schizophrenia ranges from mid to late adolescence through early adulthood; it occurs more often in males and affects them more severely than it does females (3). Although minor physical anomalies and subtle motor, social, or cognitive impairments are often observed during the premorbid stage of the illness (4, ,5), these differences generally fail to place individuals outside the normal range of functioning. In the prodromal stage, attenuated positive symptoms, mood symptoms, cognitive symptoms, social withdrawal, or obsessive behaviors may occur (5). After the onset of the full syndrome (i.e., both positive symptoms, such as delusions and hallucinations, and negative symptoms, such as impaired cognition, volition, and emotion), substantial functional deterioration (e.g., work, interpersonal relationships, or self care) typically occurs, especially during the first five to ten years, and then clinical deterioration reaches a plateau (for review, see 5). Schizophrenia is further defined by characteristic but nonspecific disturbances in the form and content of thought, perception, cognition, emotion, sense of self, volition, social relationships, and psychomotor behavior (6).
There is as yet no accepted biological validation of or laboratory test for the diagnosis of schizophrenia. The human suffering, family tragedies, and financial burden caused by schizophrenia represent a tremendous challenge for the scientific community (3). Although some insights into the etiology of schizophrenia have been developed, an understanding of the illness on the molecular level remains elusive. Molecular genetics, neuroanatomy, neurophysiology, brain imaging, and psychopharmacology thus represent important avenues for current research efforts.
Molecular Genetics of Schizophrenia
Evidence from family, twin, and adoption studies has strongly implicated genetic factors in the etiology of schizophrenia. Quantitative summaries of twin studies have consistently demonstrated the strong influence of genetic effects (7, ,8) ; a recent meta-analysis of twelve twin studies estimates the heritability of the disease at 81% (p < 0.05, 73–90%; P.F. Sullivan, K.S. Kendler, and M.D. Neale, unpublished results).
Although genetic influences on the etiology of schizophrenia are notably strong, identification of the underlying susceptibility loci has proven problematic (9). Schizophrenia is a prototypic complex trait, demonstrating non-Mendelian inheritance and important environmental influences that are reviewed in the following section. Unlike some complex traits (e.g., breast cancer or Alzheimer disease), no subtype of schizophrenia has been found to segregate in a Mendelian fashion. It is likely that there are multiple, potentially interacting genes with small effects and incomplete penetrance (10). Moreover, etiologic and locus heterogeneities are widely suspected.
linkage Studies
The difficulty of identifying specific sources of genetic variation within broad genomic regions has been a key limitation to the molecular dissection of nearly all complex traits. In schizophrenia, the numerous genomic regions that have been implicated by genome scanning pose an enormous challenge, because they are inconsistent between studies and are usually broad, sometimes containing hundreds of potential candidate genes. Nevertheless, as for other complex traits, there has been considerable recent progress for schizophrenia.
Two genes identified in linkage disequilibrium–based fine-mapping work following genome scans appear to be promising candidate genes for schizophrenia. DTNBP1 (dystrobrevin-binding protein 1, or dysbindin) was identified in a study of 270 Irish high-density schizophrenia pedigrees (11–,13). Dysbindin is a component of the dystrophin protein complex in postsynaptic densities in the brain and appears to play a role in synaptic plasticity and signal transduction. For example, the dystrophin protein complex regulates nicotinic receptor clustering and recruits specific signaling molecules, such as neuronal nitric oxide synthase, and also interacts with postsynaptic density proteins involved in N -methyl-d -aspartate (NMDA) receptor clustering (14). Thus, defects in dysbindin function could affect synaptic transmission and postsynaptic receptor regulation during development, adulthood, or both (11).
A second gene identified via follow-up of a genome scan is neuregulin 1 (NRG1), initially demonstrated in an Icelandic sample (15) and subsequently in a Scottish sample (16). Neuregulin 1 is an interesting albeit complex candidate gene for schizophrenia given its important roles in neuronal migration and brain development, and its close associations with several neurotransmitter systems [e.g., NMDA, glutamate, and γ -aminobutyrate (GABA) (15)].
The DISC-1 and DISC-2 (DISRUPTED IN SCHIZOPHRENIA) genes are additional candidates for susceptibility to schizophrenia. In a large Scottish pedigree with a high rate of schizophrenia (forty-seven percent) and related psychiatric disorders, a balanced 1:11 translocation disrupts DISC-1 and DISC-2, both genes residing at 1q42.1 (17). The functions of DISC-1 and DISC-2, however, remain elusive. In a study of Finnish families, Ekelund and colleagues presented evidence in support of the linkage of region 1q42 to schizophrenia (18), although no major locus could be identified on chromosome 1q in a large multicenter sample (19).
Several lines of research have also focused interest on chromosome 22q11. People with “22q11 deletion syndrome,” also known as DiGeorge syndrome, or velocardiofacial syndrome, have very high rates of schizophrenia (twenty-five to thirty percent) (20). Although it has been difficult to determine which of the specific genes in this genomic region may mediate 22q11 deletion syndrome, there are several intriguing candidates, such as the gene encoding COMT, the postsynaptic enzyme that metabolizes released dopamine.
Association Studies
A simpler approach to investigating genetic variation in schizophrenia is to compare specific candidate genes from patients and well-matched control subjects. Although this approach is controversial (particularly in neuropsychiatry) for its capacity to yield false positive findings (21), hundreds of association studies have yielded support for at least three genes that increase risk for schizophrenia. Each of the three genes appears to confer a relatively small increase in risk, which necessitates large sample sizes in order for individual studies to attain sufficient statistical power. The gene that encodes the type 2A serotonin receptor (HTR2A) has been implicated in a meta-analysis of twenty-eight published reports; the type 3 dopamine receptor gene (DRD3) has been similarly implicated from forty-eight published reports (22). A third gene (COMT), encoding catechol-O -methyl transferase, although not significantly associated with schizophrenia in a meta-analysis (22), nevertheless stands out in recent reports as contributing to the etiology of schizophrenia (23, ,34). The relevance of these genes to the etiology of schizophrenia is discussed below (see Neurotransmitter Hypothesis).
Microarray Analysis
Microarray analysis to assess gene expression levels in the postmortem brains of schizophrenics may identify candidate genes without regard to preconceptions of disease mechanism. For example, Hakak and colleagues (25) found in the dorsolateral prefrontal cortex of twelve schizophrenics downregulation of the genes that encode MAL (myelin and lymphocyte protein), 2’,3’-cyclic nucleotide 3’-phosphodiesterase, myelin-associated glycoprotein, transferrin, gelsolin, and the neuregulin receptor Her3 (i.e., ErbB3), all of which are involved in myelination and/or oligodendrocyte function. In another microarray study of the schizophrenic prefrontal cortex, the expression of genes encoding proteins that regulate presynaptic secretory function was compromised; the affected gene products included N-ethylmaleimide-sensitive factor and synapsin II (26). Expression of the gene encoding G protein–signaling regulator 4 (RGS4), which can regulate the duration of G protein–mediated intracellular signaling, was also found to be reduced (27). Interestingly, RGS4 maps to 1q21-22, a chromosome region that has been implicated in schizophrenia by linkage (28).
Environmental Risk Factors for Schizophrenia
Although genetic risk factors clearly play a role in the etiology of schizophrenia, there is an abundant literature on the importance of non-genetic risk factors (Table 1⇓). For example, a large population–based study indicates that urban birth and late-winter/early-spring birth are much more closely associated with schizophrenia than is having an affected first-degree relative. Similarly, most other reported environmental risk factors, such as maternal stress and prenatal malnutrition, likely alter pre- and perinatal brain development.
Risk Factors for Schizophrenia and Their Effect Sizes
A variety of maternal infections have been implicated in the risk for schizophrenia, and it has been proposed that inflammatory responses to infection, especially responses that involve cytokines generated by the mother, placenta, or fetus, represent a common mechanism (29). Inflammatory cytokines generated in response to maternal infection can have a neurotoxic effect on developing neurons (30, ,31), and cytokines regulate neurodevelopmental processes implicated in schizophrenia, such as programmed cell death and synapse development.
A recent meta-analysis of prospective, population-based studies has demonstrated that three groups of obstetric complications are significantly related to schizophrenia: a) complications of pregnancy (e.g., bleeding, preeclampsia, diabetes, and rhesus incompatibility); b) abnormal fetal growth and development (e.g., premature birth or low birth weight, congenital malformations, small head circumference); and c) complications of delivery (e.g., uterine atony, asphyxia, emergency cesarean section) [for review, see (32) ].
Fetal hypoxic or ischemic damage to the developing brain may be a common mechanism for complications associated with preeclampsia, uterine atony, asphyxia, and emergency Cesarean section (33), although clinical and subclinical infections are associated with these complications as well. Intriguingly, premature cortical synaptic pruning has been implicated in fetal hypoxia associated with brain structural abnormalities among patients with early-onset schizophrenia (34). In addition, hypoxia or ischemia in preterm babies may cause intra- and periventricular hemorrhage, and the long-term consequences of the latter include ventricular enlargement, smaller hippocampal volumes (35), and corpus callosal abnormalities. It should be noted, however, that current methods of studying obstetric complications in schizophrenia may be reaching the limit of their usefulness (32), particularly given the generally small quantitative nature of obstetric effects.
It was recently reported that the cerebrospinal fluid of twenty-nine percent of patients with recent-onset schizophrenia or schizoaffective disorder contain human endogenous retroviral sequence [(HERV)-W], the transcription of which is up-regulated in the frontal cortex of postmortem brains of schizophrenic patients (36). This report supports previous speculation as to a viral etiology of schizophrenia, and the results suggest that the onset of the illness in some patients may be related to the transcriptional activation of certain retroviral elements in the brain.
Epidemiological studies have indicated an increased incidence of schizophrenia in the offspring of women who had been subjected to irradiation in their first trimester (37, ,38). Rhesus monkeys irradiated during the period of thalamic neurogenesis in utero demonstrate neuronal losses in specific thalamic nuclei as well as decreases in cortical neuropil and, in adulthood, exhibit the deformation and loss of thalamic tissue (39) characteristic of schizophrenic brains (40, ,41). It is notable that, prior to puberty, the fetally irradiated monkeys perform normally on working memory tasks, but, as adults, show deficits similar to those exhibited in schizophrenia (39). Other environmental risk factors for schizophrenia are shown in Table 1⇑.
The Neurotransmitter Hypothesis of Schizophrenia
There are a number of theories of schizophrenia, dominated for many years by neuropharmacology, that implicate aberrant neurotransmission systems—in particular, aberrant dopaminergic, serotoninergic, and glutamatergic systems. It is unclear, however, to what extent any neurochemical findings reflect primary rather than secondary pathology, compensatory mechanisms, or environmental influences.
The Dopamine Hypothesis
The classical “dopamine hypothesis of schizophrenia” postulates a hyperactivity of dopaminergic transmission at the dopamine D2 receptor in the mesencephalic projections to the limbic striatum (42, ,43). This hypothesis remains the preeminent neurochemical theory, despite several limitations [for review, see (44) ]. The notion was initially supported by a tight correlation between the therapeutic doses of conventional antipsychotic drugs and their affinities for the D2 receptor (45, ,46). In addition, indirect dopamine agonists (e.g., l -dopa, cocaine, and amphetamines) can induce psychosis in healthy subjects and, at very low doses, provoke psychotic symptoms in schizophrenics (43). The dopamine hypothesis has received support from postmortem and positron emission tomography (PET) indications of increased dopamine D2 receptor levels in the brains of schizophrenic patients (Table 2⇓) (47). However, it has been suggested that upregulation of D2 receptor expression may be the result of adaptation to antipsychotic drug treatment rather than a biochemical abnormality intrinsic to schizophrenia. In fact, some PET studies show no significant difference in D2 receptors densities between neuroleptic-naive schizophrenics and healthy controls (48).
Summary of Neurochemical Findings in Schizophrenia
There is emerging evidence for a presynaptic dopaminergic abnormality in schizophrenia, implying dysfunction in presynaptic storage, vesicular transport, release, reuptake, and metabolic mechanisms in mesolimbic dopamine systems (49). It has been further hypothesized that dysregulation and hyper-responsiveness of presynaptic dopamine neurons could lead to lasting consequences through the induction of sensitization and/or oxidative stress (5, ,50). On the contrary, the functional activity of dopamine may be decreased in the neocortex in schizophrenia, which could be, at least partially, associated with negative symptoms (e.g., emotional or cognitive impairment) (5). Whether a dopamine hyperfunction or hypofunction occurs under minimal stress remains an open question.
Considerable data suggest that heritable abnormalities of prefrontal dopamine function are prominent features of schizophrenia that may relate to a unique role for COMT in dopamine-mediated prefrontal information processing in working memory [for review, see (51) ]. COMT inhibitors can improve working memory in both rodents (52) and humans (53). Interestingly, studies of COMT-deficient mice have demonstrated that dopamine levels are increased in the prefrontal cortex but not in the striatum, and that memory performance is enhanced (54).
Recently, Egan and coworkers have demonstrated that a COMT polymorphism that results in a valine residue at a position alternatively possessed by a methionine residue occurs at higher rates in both schizophrenics and their unaffected siblings (23). Moreover, patients and siblings containing the valine allele, which results in a COMT enzyme that is fourfold more active than the methionine allele, performed relatively poorly on a neuropsychological test of working memory and manifested inefficient brain activation as assessed by functional magnetic resonance imaging (fMRI). These findings suggest that the (high-activity) COMT valine allele impairs prefrontal cognition and physiology, and by virtue of this effect, may increase risk for schizophrenia.
The Serotoninergic System
Recent attention has focused on the involvement of serotonin (5-HT) in the pathophysiology of schizophrenia [for review, see (55) ]. The “serotonin hypothesis of schizophrenia” is informed by several observations: a) serotonin receptors are involved in the psychotomimetic and psychotogenic properties of hallucinogens [e.g., lysergic acid diethylamide (LSD)]; b) the number of cortical 5-HT2A and 5-HT1A receptors is altered in schizophrenic brains (Table 2⇑); c) 5-HT2A and 5-HT1A receptors play a role in the therapeutic and/or side-effect profiles of atypical antipsychotics (e.g., clozapine); d) certain polymorphisms of the 5-HT2A receptor gene are associated with schizophrenia; e) the trophic role of serotonin in neurodevelopment may be usurped in schizophrenia; f) 5-HT2A receptor–mediated activation of the prefrontal cortex may be impaired in some schizophrenics; and g) serotoninergic and dopaminergic systems are interdependent and may be simultaneously affected in schizophrenia (55, ,56).
The Glutamatergic System
Phencyclidine (PCP) and ketamine, both potent non-competitive antagonists of the NMDA subtype of glutamate receptor (NMDA-R), induce schizophrenia-like symptoms in healthy individuals and worsen some symptoms in schizophrenia (103, ,108). Decreased NMDA-R function may thus be a predisposing or causative factor in schizophrenia (57, 59, ,60). One of the features that distinguish NMDA-R antagonists from other psychotogenic drugs such as amphetamine and LSD is the degree to which they produce frontal cognitive deficits that mimic schizophrenia [for review, see (61) ]. Postmortem studies of schizophrenics additionally indicate abnormalities in pre- and postsynaptic glutamatergic indices (Table 2⇑). NMDA-R hypofunction in the cortical association pathways could be responsible for a variety of cognitive and other negative symptoms (62) and, in mice, partial deletion of the NMDA-R1 (NR1) subunit causes the same behavioral abnormalities as PCP (63). In addition, the NR1 hypomorphic animals manifest reduced [14 C]-2-deoxygluose uptake in the medial prefrontal and anterior cingulate cortices, as is observed in chronic schizophrenic patients (64).
The existence of anatomical and functional interrelationships between dopamine and glutamate systems in the central nervous system suggests that inhibition of the NMDA-R would influence dopamine neurotransmission (46, 65, ,66). For example, NMDA-R antagonists decrease corticofugal inhibition of subcortical dopamine neurons [for review, see (62) ] and thereby enhance the firing rate of dopamine neurons (67, ,68). In humans, PET studies of dopamine receptor occupancy after acute administration of ketamine suggest that the NMDA-R antagonists increase dopamine release in the striatum (69–,71). In contrast, chronic administration of NMDA-R antagonists elicits decreased dopamine release (69) or hypoactivity of dopamine in the prefrontal cortex (60). Kapur and Seeman (72) have recently shown that both PCP and ketamine have direct effects on D2 and 5-HT2 receptors. It has also been proposed that NMDA-R antagonists can cause an excess compensatory release of glutamate that can overactivate unoccupied non-NMDA glutamate receptors, including α -amino-3-hydroxy-5-methy-isoxazole-4-propionic acid (AMPA) and kainate receptors (73). The release of glutamate in response to NMDA-R antagonists might in part be responsible for their behavioral effects. Finally, NMDA-R hypofunction may also produce abnormalities in the neuroplasticity of neurons by altering synaptic connectivity, as discussed below.
Neurodevelopmental Hypothesis of Schizophrenia
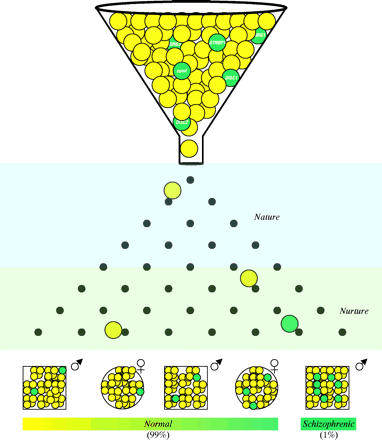
Recently, sophisticated clinical studies, advanced imaging techniques such as MRI, and novel neuroanatomical markers have provided reliable evidence that schizophrenia is a neurodevelopmental disorder (74). The “neurodevelopmental hypothesis of schizophrenia” posits that abnormalities of early brain development increase the risk for the subsequent emergence of clinical symptoms (Figure 1⇓) (75). The evidence that abnormal brain development contributes to schizophrenia comes from several domains, including: a) abnormalities of early motor and cognitive development and histories of obstetrical adversity; b) absence of evidence of neurodegeneration in postmortem tissue studies; and c) association of developmental pathological conditions with adult emergence of psychosis and related phenomena in animal and neurological models (75, ,77). In this neurodevelopmental model, schizophrenia may be associated with a subtle, static brain lesion that is caused by a combination of genetic and/or early environmental factors and that eventually interacts with normal maturational processes of the brain to facilitate symptoms such as psychosis (32). The relatively subtle childhood abnormalities (i.e., prodromal symptoms that are recognized retrospectively) may thus be a period-dependent expression of aberrant neural networks.

A general model integrating the concepts of schizophrenia After Ho and Andreasen (133).
Schizophrenia is often associated with a progressive decline in functioning. Lieberman (78) has hypothesized that a limited neurodegenerative process, most active in the early stages of the illness, may be involved in the pathophysiology of schizophrenia, and that the pathological process may be reflected by psychotic symptoms. During the course of schizophrenia, psychotic symptoms themselves may “sensitize” the brain and increase the pathology (79). Thus, it may be useful to consider what kinds of pathogenic processes might continue to compromise abnormal neural circuits.
LaMantia and colleagues have proposed that schizophrenia may involve a two- or three-“hit” process (80). In those who are diagnosed with schizophrenia later in life, a lesion occurring in early neurodevelopment (first hit), as a result of genetic load, adverse embryonic events, or perinatal events, may affect certain brain structures such as the left hippocampus and left planum temporale. This first hit would be followed by a second and possibly a third hit arising from hormonal events (e.g., altered neurosteroid biosynthesis), excitotoxicity, or oxygen radical formation so as to result in further neurodegeneration of other brain regions (81, ,82). If NMDA-R hypofunction is central to schizophrenia, resulting in excess stimulation and excitotoxic neuronal degeneration, strategies aimed at directly correcting glutamatergic dysfunction could prove to be neuroprotective against the otherwise deteriorating course of illness (61). MRI allows researchers to test these theories and identify brain regions particularly susceptible to progressive neural changes that may signify discrete stages illness (83).
At odds with the “neurodegenerative hypothesis,” schizophrenic brains do not typically manifest loss of cortical neurons, gliosis, or neuronal degeneration (84, ,85). The pathophysiology and etiology of schizophrenia may thus be related to a developmental process (e.g., neurite formation, synaptogenesis, neuronal pruning, and apoptosis), rather than a neurodegenerative process (76, ,86). Jarskog et al. (87) found low levels of the pro-apoptotic protein Bcl-2 in the temporal cortex of patients with schizophrenia, suggesting that decreased “synaptic apoptosis” may play a role in the disease.
Immunocytochemical and ultrastructural studies of postmortem brains have consistently described cellular aberrations in schizophrenia, such as decreased neuronal size, increased cellular packing density, and distortions in neuronal orientation [for review, see (88) ]. The cytoarchitectural abnormalities, such as neuronal disarray, heterotopias, and malpositioning, suggest disruption of proliferation or migration at the gestational period. Accordingly, schizophrenic brains exhibit a thirty to fifty percent reduction in the expression of reelin, a glycoprotein that acts as a “stop” signal for neuronal migration during development [for review, see (89) ] in the prefrontal cortex and hippocampus. During brain development, reelin can regulate cortical pyramidal neurons, interneurons and Purkinje cell positioning, and/or trophism. In adult brains, reelin is secreted preferentially by cortical GABAergic interneurons (81). It is intriguing that the uptake and release of GABA, the density of GABA transporter, and the level of a major enzyme in GABA biosynthesis, glutamic acid decarboxylase, have been reported to be reduced in the brain of schizophrenic patients (90–,93). If schizophrenic patients have reduced GABAergic function, their ability to inhibit increased glutamatergic activity might be deficient, thus making them more susceptible to excitotoxicity (61).
Many studies of schizophrenic brains also show low neuropil levels, abnormalities in synaptic, dendritic, axonal, and white matter tract organization, and abnormalities of glutamatergic neurotransmission, which are consistent with defective connectivity between brain regions, including the midbrain, nucleus accumbens, thalamus, temporo-limbic, and prefrontal cortices (59, 88, 94, ,95). In addition, changes in adhesion molecules (e.g., neural cell adhesion molecule), cytoskeletal proteins, neurotrophins (e.g., brain-derived neurotrophic factor), and other cell–cell signaling molecules have been observed in the brains of schizophrenic patients [for review, see (80, ,96) ].
Brain Imaging Studies of Schizophrenia
Neuroimaging techniques have revolutionized the study of schizophrenia. Findings dating from the 1970s have demonstrated that there are clear brain structural abnormalities associated with the condition (97). Structural studies performed on the postmortem brains of schizophrenics reveal many areas to be abnormal, the most robust findings being ventricular enlargement and loss of temporal lobe gray matter, although the disordered lives of the patients may admittedly involve extensive medication and dietary abnormalities that may themselves alter brain anatomy [for review, see (98, ,99) ]. A number of other changes have also been reported, but with less consistency.
Recent MRI research has identified brain characteristics associated with the early stages of psychosis. At first-episode psychosis, patients already show a range of structural brain changes, including deficits in temporal lobe (i.e., gray matter, superior temporal gyrus, and hippocampus), reduced whole brain volume, reduced prefrontal cortical gray matter volume, significant enlargement of lateral and third ventricles, and reduced thalamic volume (100–,103). Because these abnormalities exist early in the illness, they cannot arise from artifacts due to medication, trauma, or diet. Structural or functional defects of the thalamus, because it is associated with filtering sensory information and gating mechanisms, may be particularly important in the pathogenesis of schizophrenia; indeed, problems of information filtering may underlie disturbed thought processes (102, ,104). Although some PET studies suggest low activity levels in the thalamus of schizophrenic subjects (105–,109), other data suggest elevated activity in schizophrenia (110). In patients with first-episode schizophrenia, volume reduction of the superior temporal gyrus, including bilateral Heschl’s gyrus gray matter and left planum temporale gray matter, may provide an anatomical basis for the pathophysiological mechanisms that give rise to deficits in language and thought processing in schizophrenia (100).
A recent series of MRI studies has generated enthusiasm for a “neurodegenerative hypothesis of schizophrenia” that posits that there may be destruction of neural tissue associated with psychosis (78, 86, 111, ,112). Supporting this position, a recent controlled longitudinal MRI study of chronic schizophrenia demonstrated accelerated frontotemporal cortical volume decline (83). Interestingly, greater clinical severity was associated with faster rates of the brain volume changes. It is, however, hard to imagine how the magnitude and duration of changes observed in MRI studies of schizophrenia could be occurring as a neurodegenerative process, whether by cell necrosis or apoptosis, without observable evidence of neuronal loss and other related changes in postmortem tissue (113).
Studies using fMRI have identified neural structures associated with observed cognitive problems, and show cortical responses in relation to psychopathological aspects of schizophrenia. Specifically, fMRI analysis of working memory tasks (e.g., two-back or continuous performance test) reveals reduced activity in frontal areas of the brain such as the dorsolateral prefrontal cortex (114, ,115) and the anterior cingulate cortex (116, ,117). Some fMRI studies also suggest that auditory hallucinatory states are associated with activation in the inferior and frontal insular, anterior cingulate, bilateral temporal cortex (with greater responses on the right), right thalamus, inferior colliculus, left hippocampus, and parahippocampal cortex (118, ,119). Silbersweig and colleagues (120), using PET, reported similar results.
Magnetic resonance spectroscopy (MRS) has been increasingly used to measure in vivo metabolite levels in particular regions of the brain in schizophrenia, although it has less spatial resolution [for review, see (121–,123) ]. The results of 31 P-MRS suggest decreased synthesis and increased degradation of membrane phospholipids in prefrontal cortical regions and medial temporal lobe structures at certain phases of schizophrenia. Some, but not all, 1 H-MRS studies also indicate a decrease in neuronal cell mass in the hippocampal region and the frontal lobes, which supports direct volumetric studies. In addition, 1 H-MRS studies of schizophrenics show increased levels of glutamine and glutamate in frontal lobe voxels, which are associated with illness duration and reduced by atypical antipsychotic drug treatment (115).
Developing Novel Antipsychotic Drugs
In recent years, concerted research and development efforts have sought novel “atypical” antipsychotic drugs, offering the therapeutic advantages of clozapine without the associated risk of blood dyscrasias, and have yielded risperidone, olanzapine, quetiapine, ziprasidone, sertindole, sulpiride, amisulpride, zotepine, aripiprazole, and iloperidone. Relative to conventional neuroleptics, many new medications offer the advantages of improved negative symptoms and cognitive impairment, fewer extrapyramidal symptoms, and less tardive dyskinesia. The advent of atypical antipsychotics represents the first significant advance in the pharmacological treatment of schizophrenia in the past four decades. We are, however, still in the process of defining the exact nature and clinical profiles of atypical drugs in terms of their therapeutic efficacy and adverse effects.
A number of partial agonists of the D2 receptor are currently in clinical trials and seem to offer promise [for review, see (124) ]. Drugs of this class, including 3-(3-hydroxyphenyl)-N -propylpiperidine, could stabilize dopaminergic tone, that is, they are capable of alleviating signs of hyperdopaminergia without reducing dopaminergic function below the baseline level (62). Aripiprazole (OPC-14597) is a dopamine autoreceptor partial agonist and postsynaptic D2 receptor antagonist, and has modest affinity for 5-HT1A, 5-HT2A, 5-HT6, and 5-HT7 receptors (125). Aripiprazole appears to be the first clinically effective antipsychotic partial D2 agonist.
A number of agents that interact directly with the glutamatergic system are currently in development and eagerly awaited (62). Examples of the new “glutamate-based” agents are the glycine site agonists (e.g., glycine, d -serine, d -cycloserine), glycine reuptake inhibitors, glutamate release inhibitors (e.g., LY-354740 and lamotrigine), AMPA agonists and antagonists (e.g., LY-293558 and GYKI 52466), and ampakines (e.g., CX-516) [for review, see (61) ]. The glycine site agonists appear to be effective in reducing negative symptoms and cognitive impairment when added to ongoing antipsychotic treatment (with the exception of clozapine) in patients with schizophrenia. Their beneficial effects on positive and depressive symptoms are less robust. Of these glycine agonists, d -serine appears to be the most promising agent [for review, see (61, ,126) ]. In the case of AMPA ligands, it seems unclear if agonists, antagonists, or partial agonists/modulators will be most successful. Lastly, drugs acting on different subtypes of metabotropic glutamate receptors (e.g., LY-354740) seem to offer some promise (162).
There is a large body of anatomical and pharmacological evidence indicating that cholinergic muscarinic receptors may modulate dopamine and glutamatergic neurons [for review, see (127) ]. Moreover, the fact that schizophrenics have sensory gating and cognitive dysfunction suggests a role for the cholinergic system in the etiology and therapy of these deficits. Partial agonists of muscarinic receptors that indicate antipsychotic activity in animal models may find utility in the treatment of schizophrenia. Examples of these agents are the muscarinic M1 /M4 agonist xanomeline and the muscarinic M2 /M4 agonists PTAC [(5R,6R) 6-(3-propylthio-1,2,5-thiadiazol-4-yl)-1-azabicyclo[3.2.1]octane] and BuTAC [(5R,6R) 6-(3-butyllthio-1,2,5-thiadiazol-4-yl)-1-azabicyclo[3.2.1]octane] [for review, see (127) ]. Xanomeline has been demonstrated to have positive effects on cognitive and psychotic-like symptoms (e.g., hallucinations and delusions) in Alzheimer’s disease (128). Accumulating data suggest that muscarinic partial agonists might be efficacious in treating not only positive, but also negative and cognitive symptoms in schizophrenia (129, ,130).
Future Directions
The tools of modern neuroscience, drawing from neuroanatomy, neurophysiology, brain imaging, genetic analysis, and psychopharmacology, promise to provide a host of new insights into the etiology and treatment of schizophrenia. Increasing knowledge of pathophysiology and better characterization of genotype–phenotype relationships may lay the way toward rational drug design and the identification of environmental factors that pose risks for individuals. Conversely, our abilities to identify high-risk individuals will contribute to realistic preventive measures.
Current data suggest that schizophrenia may represent a spectrum of phenotypic consequences that overlie a group of disorders whose etiopathogenesis involves the interplay of complex polygenic influences and environmental risk factors operating on brain maturational processes. Clearly, the complexity of these potential gene–environment–development interactions presents a tremendous challenge for the clinical elaboration of mechanisms operative in schizophrenia. The endgame in uncovering the etiology of complex, polygenic disorders will depend on clarification of allele function in health and illness. In the coming decade, we may see the first reports of studies that examine precisely measured genetic and environmental causes of schizophrenia for a given population (131). We will also have to take into account the dynamic interplay between genes and environment in utero. Animal models of schizophrenia that focus on developmental events will be crucial for assessment of neurobiological processes that occur early in the illness (39). Hypothesis-independent approaches, such as linkage and gene expression profiling may simplify gene identification.
Structural and functional brain imaging suggests both global and regional abnormalities as well as disconnections of specific cortical-subcortical circuits (132). New approaches to study brain activity in vivo may give leads as to where to look for abnormalities, and could provide an objective index that reflects vulnerability, clinical state, and prognosis of schizophrenia (122). Imaging measurements also promise to unite pathophysiological, genetic, and etiological research efforts (122). Furthermore, the usefulness of fMRI in monitoring and evaluating neurobiological responses to psychosocial therapies must also be thoroughly explored. Recent developments in profiling gene expression (e.g., gene chips) may allow rapid screening of novel antipsychotic drugs and the pharmacogenetic ramifications of their side effects. Future research must also consider events upstream as well as downstream to receptors, and attention must be directed to the complete range of neurotransmitters, including acetylcholine, GABA, neuropeptides, and neurosteroids.
Conclusion
The conclusive identification of specific etiological factors and pathogenic processes in schizophrenia remains an urgent goal. In addition to improving prognoses, studies of the illness may more generally deepen our understanding of the normal human brain and mind. Biological markers for schizophrenia may come from a combination of assessment techniques, with imaging techniques adding the crucial ingredient for high-resolution structural and functional assessments of brain regions. Indeed, opportunities for schizophrenic patients and their physicians to cope with the disease are better than ever before.
Acknowledgments
This work was supported in part by the UNC Schizophrenia Research Center, an NIMH Silvio O. Conte Center for the Neuroscience of Mental Disorders (MH064065), and the Foundation of Hope of Raleigh North Carolina. The authors would like to acknowledge the support of Janssen Pharmaceutical K.K. (Japan) in preparing this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2003

