BLOOD-BRAIN BARRIER DRUG TARGETING: THE FUTURE OF BRAIN DRUG DEVELOPMENT
Abstract
The development of new drugs for the brain has not kept pace with progress in the molecular neurosciences, because the majority of new drugs discovered do not cross the blood-brain barrier (BBB). Although approximately 100% of large-molecule drugs do not cross the BBB, the problem is nearly as severe for small-molecule drugs—greater than 98% of small-molecule drugs do not cross the BBB. Despite this situation, no pharmaceutical company in the world today has a BBB drug-targeting program. Nevertheless, BBB drug targeting technology can be built around a knowledge base of the endogenous transporters within the brain capillary endothelium, which forms the BBB in vivo.
Introduction
The global market for drugs for the central nervous system (CNS) is greatly underpenetrated and would have to grow by over 500% just to be comparable to the global market for cardiovascular drugs (1). The principle reason for this under-development of the global brain drug market is that the great majority of drugs do not cross the brain capillary wall, which forms the blood-brain barrier (BBB) in vivo. Only a small class of drugs—small molecules with high lipid solubility and a low molecular mass (Mr ) of < 400–500 Daltons (Da)—actually cross the BBB (2) . However, there are only a few diseases of the brain that consistently respond to this category of small molecules (3, 4), and these include depression, affective disorders, chronic pain, and epilepsy. In contrast, many other serious illness disorders of the brain do not respond to the conventional lipid-soluble–low-Mr small-molecule therapeutics (1) , and these include Alzheimer disease, stroke/neuroprotection, brain and spinal cord injury, brain cancer, HIV infection of the brain, various ataxia-producing disorders, amytrophic lateral sclerosis (ALS), Huntington disease, and childhood inborn genetic errors affecting the brain. To this latter list one could also add Parkinson disease (PD) and multiple sclerosis (MS). Although l -dihydroxyphenylalanine (l -Dopa) therapy has been available for decades to treat PD, there has been no neuroprotective drug for PD that halts the inexorable neurodegeneration of this common disorder. Although patients with MS have benefited from cytokine drug therapy, which acts on the peripheral immune system, there is no drug that stops the inevitable demyelination within the CNS caused by MS. Many, if not most, of the CNS disorders that are refractory to small-molecule drug therapy might be treated with large-molecule drugs including recombinant proteins and gene-based medicines; however, several hindrances, biochemical and economic, are inhibiting their development.
Rate-Limiting Role of the BBB in Brain Drug Development
Present-day incongruities in brain drug development are illustrated by a consideration of some of the characteristics of the CNS drug industry. Whereas 98% of all small-molecule drugs do not cross the BBB, and nearly 100% of large-molecule drugs do not cross the BBB, none of the global pharmaceutical companies have a BBB drug-targeting program.
The Limitations of Small–Molecule Drugs
Drug companies today do not have in-house BBB drug targeting programs because it is widely believed that (a) most disorders of the brain respond to small molecules and (b) most small molecules cross the BBB. However, only a few brain diseases (as described above) consistently respond to lipid-soluble small molecules (3, 4). This fact is illustrated by several reviews of current CNS drugs. In one study of the comprehensive medicinal chemistry (CMC) database (4) , over 7,000 drugs were analyzed and only 5% of these drugs affected the CNS, and these CNS-active drugs treated only depression, schizophrenia, and insomnia. The average Mr of the CNS active drug was 357-Da. In another study, only 12% of drugs were active in the CNS and only 1% of the total number of drugs were active in the CNS for diseases other than affective disorders (5) .
The other problem with small molecules is that only a small percentage of them cross the BBB in pharmacologically significant amounts. The rate-limiting role of the BBB is illustrated with histamine, a small molecule of only 111 Da. Histamine, however, does not cross the BBB, and the inability of histamine to penetrate the brain is illustrated in Figure 1A⇓ (6) . Histamine has too many hydrogen-bond-forming functional groups, and BBB penetration is inversely related to the number of hydrogen bonds that a drug forms with solvent water (2) . Molecules that do cross the BBB typically are lipid soluble and have a Mr threshold of 400-500 Da.
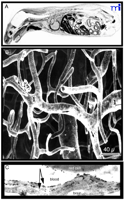
Structure of the Blood–Brain Barrier (BBB). A.
Autoradiograph of a mouse taken 30 minutes after intravenous injection of radiolabeled histamine, a small molecule that does not cross the blood–brain barrier. The drug is taken up by all organs of the body except the brain and spinal cord. Reprinted with permission (6). B. Scanning electron micrograph of the vascular cast of the human cortical microvasculature. The capillaries are separated by a distance of approximately 40 μm. Thus, each neuron is virtually perfused by its own blood vessel. Reprinted with permission (10). C. Immunogold electron micrograph of the capillary endothelium of the human brain stained with an antibody specific for the GLUT1 glucose transporter. The transporter is found on the erythrocyte plasma membrane, on the luminal membrane of the capillary endothelium, which is the blood side of the BBB, and on the abluminal membrane of the capillary endothelium, which is the brain side of the BBB. A distance of approximately 300 nm separates the luminal and abluminal membranes of the capillary endothelium (arrows). Reprinted with permission (59).
Virtually all drugs developed from receptor-based high throughput–screening (HTS) programs for CNS drug discovery are either water soluble with a high degree of hydrogen bonding or have a Mr greater than 400-500 Da. With the introduction of HTS-based CNS drug discovery, the Mr of the drugs has increased and the lipid solubility of drugs has decreased (5) . Without a parallel effort in CNS drug-targeting, virtually all HTS-based CNS drug discovery programs will invariably end in program termination. Large-molecule drugs are not developed for the brain because of the BBB problem. Indeed, if a large-molecule drug is found to be effective for the brain, the molecule is generally abandoned and a search is initiated for a small-molecule peptidomimetic. However, with the exception of those situations where the endogenous ligand is itself a small molecule, no small-molecule peptidomimetics have been discovered to date that are capable of transport across the BBB. Therefore, the small-molecule peptidomimetic will still have to be reformulated to enable BBB transport, and the development of a small-molecule BBB drug-targeting strategy can just as challenging as the development of a large-molecule BBB drug-targeting strategy.
Craniotomy-Based Brain Drug Delivery
There are examples of CNS drug development programs that go forward even though it is known that the drug does not cross the BBB and that no BBB drug delivery strategy is available. In this setting, the strategy for dealing with the BBB problem is to administer the drug after drilling a hole in the head, a process called craniotomy. With this approach, the small- or large-molecule drug may be administered either by intracerebroventricular (ICV) or intracerebral (IC) injection. With IC administration (7) , the drug stays at the depot site at the tip of the injection needle or at the margins of the polymeric implant (Figure 2A⇓). With ICV administration (8), the drug only distributes to the ependymal surface of the ipsilateral ventricle and does not significantly penetrate into brain parenchyma (Figure 2B⇓). Thus, the treatment volume with either ICV or IC administration is less than 1% of the brain volume, and there are few, if any, brain diseases that are treatable with such limited penetration of drug into the brain.
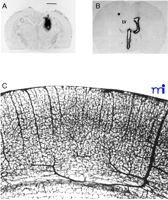
Transcranial and transvascular drug delivery to the the brain. A. Film autoradiogram of rat brain 48 hours after an intracerebral implantation of a polymeric disc containing radiolabeled nerve growth factor (NGF). The magnification bar is 2.5 mm, and the diameter of the polymeric implant is 2.0 mm. Therefore, there has been little distribution of the drug away from the polymeric implant during the 48-hour period. Reprinted with permission (7). B. Autoradiograph of rat brain 20 hours after a single intracerebroventricular injection of radiolabeled brain derived neurotrophic factor (BDNF) into the lateral ventricle (LV). The drug distributes only to the ependymal surface of the ipsilateral lateral ventricle and to the third ventricle (3V) prior to exodus from the spinal fluid compartment back to the peripheral bloodstream. There is no significant distribution of the drug to the contralateral side, and there is no significant penetration of the drug into brain parenchyma from the ependymal surface. Reprinted with permission (8). C. India ink injection study of rat brain showing the density of the cortical microvasculature. Because brain capillaries are separated by a distance of only about 40 microns, any drug that crosses the vascular barrier via the transvascular route to brain will immediately distribute to the extracellular space of the entire brain. Reprinted with permission (9).
The vascular route to the brain
In contrast to the inefficiency of craniotomy-based drug delivery to the brain, a transvascular route of drug administration, following intravenous or systemic injection, can treat virtually 100% of the neurons in the brain. The density of the microvasculature in the rat brain is illustrated in Figure 2C⇑ (9) . Because every neuron is perfused by its own blood vessel, the drug is delivered to the “doorstep” of every neuron in the brain following initial transport across the vascular barrier (Figure 2C⇑). In the human brain, there are approximately 100 billion capillaries totaling 400 miles in length (2) , and these are illustrated with the scanning electron micrograph in Figure 1B⇑ (10) . The combined surface area of brain capillary endothelium is approximately 20 m2 in the human brain (2). The delivery of drugs (or genes) to the brain by the transvascular route is so efficient that the drug or gene could be delivered to all parts of the brain once the vascular barrier is traversed. However, in the absence of a BBB drug-targeting system, the transvascular route to the brain is virtually impenetrable by the majority of drug candidates (Figure 1A⇑). If the large numbers of patients worldwide that are afflicted with serious disorders of the brain and spinal cord are to be treated, then the present trend of persistent under-development of BBB transport biology must be reversed.
Outline of a Blood-Brain Barrier Drug Targeting Program
There are both chemistry-based and biology-based approaches for developing BBB drug-targeting strategies (Figure 3⇓) (11) . The chemistry-based strategies are the conventional approaches that rely on lipid-mediated drug transport across the BBB. The limitations of lipid-mediated BBB drug transport are discussed below. The biology-based approaches (Figure 3⇓) require prior knowledge of the endogenous transport systems within the brain capillary endothelium, which forms the BBB in vivo. The biology-based strategies for brain drug delivery are founded on the principle that there are numerous endogenous transport systems within the BBB, and that these transporters are conduits to the brain. The endogenous BBB transport systems may be broadly classified as carrier-mediated transport (CMT), active efflux transport (AET), and receptor-mediated transport (RMT). These BBB transport systems are situated on the luminal and abluminal membranes of the brain capillary endothelium. For example, the expression of the Glut1 glucose transporter on both the luminal and abluminal membranes of the capillary endothelium of the human brain is illustrated in Figure 1C⇑ (12).
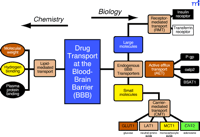
Outline of a program for developing BBB drug targeting strategies derived from either chemistry-based or biology-based disciplines. Chemistry-based strategies emphasize lipid solubility, hydrogen bonding, and molecular weight of the drug. Biology-based strategies emphasize endogenous BBB transporters. Small molecules can be transported across the BBB by either accessing certain carrier-mediated transport (CMT) systems or by inhibiting certain active efflux transporters (AET). Large-molecule drugs such as recombinant proteins or gene medicines can be delivered across the BBB via the receptor-mediated transport (RMT) systems. Reprinted with permission (11).
Drug delivery to the brain through the many endogenous transport systems within the BBB requires reformulation of the drug so that the drug can access the BBB transport system and enter the brain. The biology-based approaches to solving the BBB drug-delivery problem require advance knowledge of the endogenous transporters and could only be accomplished within the pharmaceutical industry if an in-house brain drug-targeting program was supported to the same extent as the in-house brain drug-discovery program. Researchers within brain drug-discovery and brain drug-targeting could then work closely together in the drug development process to ensure that a viable reformulation of the drug is accomplished at the earliest of preclinical stages. Thus, the dual goals of brain drug formulation are to enable BBB transport and retain the biological activity of the pharmaceutical.
Chemistry-Based Approach: BBB Lipid-Mediated Transport
There are two ways that a drug can be lipidated. First, the polar functional groups on the water-soluble drug can be masked by conjugating them with lipid-soluble moieties. Second, the water-soluble drug can be conjugated to a lipid-soluble drug carrier. Either reformulation of the drug leads to the production of a prodrug, which is lipid soluble and can cross the BBB. Ideally, the prodrug is metabolized within the brain and converted to the parent drug. Apart from the di-acetylation of morphine to create heroin (13), there have been few examples wherein the prodrug approach has been used to successfully solve the BBB drug-delivery problem in clinical practice. Two limitations of the prodrug approach are the adverse pharmacokinetics and the increased molecular weight of the drug that follow from lipidation.
The pharmacokinetic rule
The percent of injected dose (ID) of a drug that is delivered per gram brain (%ID/g) is directly proportional to both the
BBB permeability–surface area (PS) product and the area under the plasma concentration curve (AUC):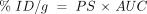
When a drug is lipidated, the BBB PS product is increased. However, the penetration of the lipidated drug is also increased in all organs of the body, which alters the plasma clearance of the drug. Following lipidation, the blood half-time of a drug may decrease from several hours to only a few minutes. Thus, there is a reduction in the plasma AUC in parallel with the increase in membrane permeation caused by lipidation. The increased PS product and the decreased plasma AUC have offsetting effects leading to nominal increases in the % ID/g of brain, which is not increased in proportion to the increase in BBB PS product or lipid solubility.
Molecular weight threshold
The conversion of a water-soluble drug into a lipid-soluble prodrug leads to an increase in the Mr of the drug. This increase in Mr can be substantial depending on the strategy used to lipidate the drug. The molecular weight of virtually all CNS-directed drugs in present-day clinical practice are under 400–500 Da (3–5). Lipid-soluble drugs with masses above the 400–500 Da threshold, with some exceptions, do not cross the BBB in pharmacologically significant amounts. The biophysical basis of the mass-specific threshold of BBB drug transport is explicable within the context of a pore model of lipid-mediated transport across biological membranes (14) . The membrane phospholipid bilayer is not inert but is mobile in living cells. This mobility causes kinks in the long chain fatty acyl groups that create transient pores within the membrane to enable “molecular hitchhiking” of the lipid-soluble small-molecule drugs across biological membranes (14) . This model would not be applicable for drug diffusion through solvents, which reinforces the idea that drug diffusion across biological membranes is not effectively modeled by drug diffusion through solvents, particularly when the molecular mass of the drug exceeds 400-Da (15). The permeation of a drug through a biological membrane decreases exponentially as the molecular size of the drug increases (16) . For BBB transport, the upper limit in molecular area appears to be about 80 Å2 , which corresponds to a Mr of less than 300–400 Da. If the size of the drug is doubled from 50 Å2 (Mr about 250-Da) to 100 Å2 (Mr about 400-Da), the BBB permeation decreases by 100-fold (16) . Thus, if the lipidation of a drug causes a significant increase in square area of the molecule, the drug may be too large to effectively cross the BBB. The fact that membrane permeation does not increase in proportion to the increase in lipid solubility when the Mr of the drug is increased has been known for more than 30 years (17) , but little research is done in this area. There are still only rudimentary models of how lipid soluble drugs physically traverse a biological membrane.
Present-day CNS drug-development programs are facing severe challenges in the discovery and development of new drugs for the many disorders of the brain. These challenges derive from (i) the extent to which the BBB limits brain uptake of virtually all drug candidates and (ii) the limitations of the traditional or chemistry-based approaches to solving the BBB problem. It may be time to consider the biology-based approaches to the BBB problem, which requires an understanding of the endogenous transport systems within the BBB (Figure 3⇑).
Biology-Based Approach: BBB Carrier-Mediated Transport
The conversion of dopamine, a water-soluble catecholamine that does not cross the BBB, into the corresponding α -amino acid, l -DOPA, enables dopamine delivery to the brain, which has been the mainstay of treatment of PD for nearly 40 years (18) . The use of l -DOPA to deliver dopamine to the brain is a BBB drug-delivery strategy that utilizes the type 1 large neutral amino acid transporter (LAT1)—one of the CMT systems within the BBB. Upon crossing the BBB through LAT1, l -DOPA is converted back to dopamine within the brain by aromatic amino acid decarboxylase (AAAD). Other drugs that cross the BBB via LAT1 include melphalan for brain cancer, α -methyl-DOPA for treatment of high blood pressure, and gabapentin for epilepsy (19–21). Apart from LAT1, there are other BBB CMT systems that could be accessed to solve BBB drug-delivery problems (Figure 4⇓), including the GLUT1 glucose transporter, the MCT1 lactate transporter, the CAT1 cationic amino acid transporter, and the CNT2 adenosine transporter, among others. If the BBB CMT systems are to be exploited to overcome the BBB drug-delivery problem, the drug must be reformulated such that the drug assumes a molecular structure mimicking that of the endogenous ligand. This principle is illustrated by gabapentin, which is 1-(aminoethyl) cyclohexaneacetic acid. Gabapentin is a γ -amino acid, not an α -amino acid. However, this drug’s structure does mimic that of an α -amino acid and is recognized by the BBB LAT1 large neutral amino acid transporter (21) . In the absence of LAT1-mediated transport across the BBB, gabapentin would be too water soluble to cross (via lipid mediation) the BBB in pharmacologically significant amounts. An alternative strategy to accessing the BBB CMT systems is to conjugate the drug to a nutrient such as glucose, which crosses the BBB on its own CMT system. However, this approach generally is not successful. The drug–nutrient conjugate will invariably not be recognized by the stereoselective pores that are formed by the individual BBB CMT transporter proteins. Rather, the structure of the drug must mimic the structure of the endogenous nutrient so that the drug can effectively bind to the active site of the BBB CMT transporter protein.
BBB transport systems. Processes involved in ferrying molecules across the BBB include carrier-mediated transport (CMT), active efflux transport (AET), and receptor-mediated transport (RMT). Examples of CMT systems include the GLUT1 glucose transporter, the LAT1 large neutral amino-acid transporter, the CAT1 cationic amino-acid transporter, the MCT1 monocarboxylic acid or lactate transporter, and the CNT2 adenosine transporter. AET, in the brain-to-blood direction, involves the sequential action of an energy-dependent transporter and an energy-independent exchanger at opposite poles of the capillary endothelium. Examples of the energy-dependent systems include P-glycoprotein (Pgp) and the multidrug resistance proteins (MRPs). Examples of the sodium-independent exchangers include organic anion-transporting polypeptide type 2 (oatp2) and BBB-specific anion transporter type 1 (BSAT1), also known as oatp14. The BBB RMT systems include the insulin receptor (IR), the transferrin receptor (TfR), the insulin-like growth factor receptor (IGF-R), the leptin receptor (OB-R), the neonatal Fc receptor (FcRn), or the type BI scavenger receptor (SR-BI). The BBB TfR is located on both luminal and abluminal membranes and mediates the bi-directional transport of transferrin across the BBB. The FcRn is selectively localized on the abluminal membrane and mediates the asymmetric efflux of immunoglobulin G (IgG) molecules from brain to blood. The SR-BI is selectively localized on the luminal membrane of the capillary endothelium and mediates the endocytosis of modified lipoproteins from blood into the brain capillary endothelial compartment, without significant transcytosis through the endothelial barrier.
The BBB CMT systems are generally equilibrative transporters that mediate the blood-to-brain and brain-to-blood transport of the nutrient in either direction across the BBB, owing to the expression of the CMT system on both the luminal and abluminal membranes of the brain capillary endothelium (Figures 1C and 4⇑⇑). An exception to this rule is the adenosine transporter, CNT2, which is partially sodium dependent (22) . The transport of adenosine from blood to brain is also characterized by an “enzymatic BBB,” which blocks the uptake of circulating adenosine into brain interstitial fluid (23) . Although there is an adenosine CNT2 transporter at the BBB on the luminal membrane of the capillary endothelium, there is no increase in cerebral blood flow following intracarotid arterial infusion of adenosine (24) . Once the adenosine enters the intra-endothelial compartment, the molecule is rapidly metabolized, and little free adenosine escapes across the abluminal membrane into brain interstitial fluid.
Enzymatic BBB
The different components of the “enzymatic BBB” (25) must be considered in addition to the endogenous BBB transport systems when designing brain drug delivery strategies. The enzymatic systems that degrade molecules crossing the endothelial membrane may be expressed on the endothelial plasma membrane, the pericyte plasma membrane, or the astrocyte foot process. The brain capillary endothelial cell and the brain capillary pericyte, which sits on the brain-side of the endothelium, share a common microvascular basement membrane. Nearly 100% of the surface area of the capillary basement membrane is covered by end-feet of processes originating from brain astrocytes, and these astrocytic end-feet are separated from the capillary endothelium by a distance of only 20 nm. In fact, the endothelium, the pericyte, and the astrocyte foot process work in concert to tightly regulate the flux of molecules between blood and brain across the microvascular barrier (2) .
Molecular biology of BBB carrier-mediated transporters
Some of the BBB CMTs have been cloned, and from their full-length cDNAs, RNA is transcribed and prepared that can be injected into frog oocytes for the expression of BBB transporters. This methodology enables the measurement of the transport kinetics of these transporter proteins. The complementary RNA (cRNA) from BBB CNT2 is particularly active in frog oocytes and enabled the detailed kinetic analysis of the transport of dideoxyinosine (DDI) via the CNT2 transporter (26) . This molecular biological approach to BBB CMT systems is to be preferred over in vitro BBB models. Although brain capillary endothelial cells may be grown in tissue culture to form an “in vitro BBB”, the gene expression of many of the BBB CMT systems is severely downregulated in tissue culture. Indeed, the transport of l -DOPA across the BBB by the CMT (e.g., LAT1) system would probably not be detected in an in vitro BBB screen, owing to decreased gene expression of the BBB LAT1 in brain endothelium grown in tissue culture.
Biology-Based Approach: BBB Active Efflux Transport
P-glycoprotein (Pgp) is the prototypic AET system found at the BBB. However, there are many other AETs other than Pgp that function at the BBB to cause the selective export of metabolites from brain back to blood. Although Pgp is principally expressed at the capillary endothelium in rodent brains, this transporter is also expressed at both the capillary endothelium and at astrocyte processes in primate and human brains (27, 28). Within the brain capillary endothelium, it is assumed that Pgp is selectively localized at the luminal membrane, although the definitive immunogold electron-microscopic studies for this transporter have yet to be performed for brain. The GLUT1 glucose transporter is expressed at both the luminal and abluminal endothelial membranes in rat brain (29) , and this transporter comigrates with Pgp in fractionated plasma membranes from rat brain endothelia (30).
Polarity of BBB active efflux transporters
Active efflux transport at the BBB is likely the result of the concerted action of energy-dependent and energy-independent transport systems selectively localized to the luminal and abluminal endothelial membranes, similar to the polarity of glucose transporters at the apical and basolateral membranes of renal tubular epithelium (31) . Energy-independent exchangers may be expressed at the abluminal membrane, and work in conjunction with ATP-dependent transporters, such as Pgp, at the luminal membrane (Figure 4⇑). Alternatively, sodium-dependent co-transporters may be expressed at the abluminal membrane and work in concert with energy-independent exchangers at the luminal endothelial membrane. Candidates for energy-dependent active transporters at the BBB include Pgp or certain multi-drug resistance proteins (MRPs) (32).
Candidates for the sodium-independent exchangers at the BBB include organic anion–transporting polypeptide type 2 (oatp2) (33, 34) , or BBB specific anion transporter type 1 (BSAT1) (35) , which is also a member of the oatp family and is designated oatp14 (36).
Codrugs
Drugs that inhibit a BBB AET could be used as a “codrug” to cause increased brain penetration of a therapeutic drug that is normally excluded from brain by a BBB AET system. For example, AAAD inhibitors are administered as codrugs in conjunction with l -DOPA to optimize brain penetration of the l -DOPA. The discovery of codrugs that inhibit BBB AET systems would be facilitated by the initial cloning of these transporters, followed by their expression in oocytes or some alternative system to enable the development of a CNS codrug discovery program.
Active efflux (transport) of azidothymidine (AZT) across the BBB
The human immunodeficiency virus (HIV) affects the brain early in the course of the disease that ultimately progresses to acquired immune deficiency syndrome (AIDS). AZT readily crosses the choroid plexus epithelial barrier, which forms the blood-cerebrospinal fluid (CSF) barrier, and enters CSF (37) . However, AZT penetration in the brain parenchyma is minimal, owing to very restrictive transport at the BBB (38) . The AZT model illustrates that drug distribution in the CSF reflects transport across the blood–CSF barrier, not drug transport across the BBB. Drugs may readily enter CSF but might penetrate brain poorly owing to restrictive transport across the BBB. Drug entry into CSF should not be used as an index of BBB transport of the drug because the biological transport properties of the BBB and the blood-CSF barrier are different. Once the drug enters into the spinal fluid compartment via transport across the blood-CSF barrier, the drug is rapidly exported back to the peripheral circulation via absorption across the arachnoid villi into the superior sagittal sinus. This process occurs by bulk flow at rates several orders of magnitude faster than the slow diffusion of drug into brain parenchyma from the ependymal surface (2) . AZT penetration into the brain is poor because this drug is a substrate for a BBB AET system (39) . The BBB AZT active efflux transporter has yet to be characterized at the molecular level but is not Pgp (40) . The discovery of the transporter responsible for AZT active efflux across the BBB would enable the development of co-drugs that inhibit this system and increase brain penetration of AZT from blood. Virtually all of the drugs presently in clinical practice for the treatment of AIDS do not cross the BBB, owing to active efflux transport. HIV protease inhibitors are substrates for Pgp (41) , and the nucleoside reverse transcriptase inhibitors such as AZT or 3TC are substrates for non-Pgp BBB AETs. Therefore, present-day highly active antiretroviral therapy (HAART) for AIDS selectively inhibits virus replication in the peripheral tissues to a greater extent than in the CNS, which allows the brain to serve as a sanctuary for the HIV.
Biology-Based Approach: BBB Receptor-Mediated Transport
Certain endogenous large-molecule neuropeptides such as insulin, transferrin, or leptin access the brain from blood via receptor-mediated transport (RMT) across the BBB (Figure 4⇑). This transport is mediated by specialized ligand-specific receptor systems, including the insulin receptor (IR) or the transferrin receptor (TfR), which are highly expressed on the capillary endothelium of brain (2) . Certain peptidomimetic monoclonal antibodies (MAbs) bind to exofacial epitopes on the BBB receptors. These epitopes are spatially separated from the endogenous ligand-binding site, and the binding of MAbs to the BBB receptor enables RMT of the peptidomimetic MAb across the BBB in vivo. These peptidomimetic MAbs may be used as “molecular Trojan horses” to ferry large-molecule drugs (e.g., recombinant proteins, gene-based medicines) across the BBB (2) .
BBB Transport of Recombinant Proteins
Recombinant Proteins as Neurodiagnostics
Many human brain cancers overexpress the receptor for epidermal growth factor (EGF). Radiolabeled ligands of the EGF receptor, such as EGF itself, could be used as peptide radiopharmaceutical imaging agents for the early detection of brain cancer. However, EGF does not cross the BBB, even in brain tumors (42) . Because of the BBB problem, EGF cannot be developed as a peptide radiopharmaceutical for neuroimaging. Similarly, none of the hundreds of other endogenous neuropeptides can be developed as receptor-specific peptide radiopharmaceuticals for neuroimaging because these molecules do not cross the BBB. Present day neuroimaging is limited to a few lipid soluble small molecules that access one of a few monoaminergic or amino acidergic neurotransmission systems. However, the number of peptidergic neurotransmission systems in the brain is nearly two orders of magnitude greater than the number of small-molecule neurotransmission systems. The potential for neuroimaging would be increased if neuropeptide radiopharmaceuticals could be reformulated to enable BBB transport.
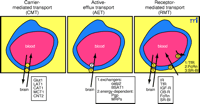
The molecular reformulation of EGF to enable BBB transport (Figure 5A⇓) involves conjugation of the EGF to a BBB molecular Trojan horse consisting of a monoclonal antibody (MAb) to the transferrin receptor (TfR). The TfR MAb acts as a molecular Trojan horse to ferry drugs across the BBB because the brain capillary endothelium is enriched in TfR (Figure 4⇑). The TfR MAb–EGF conjugate binds the BBB TfR and is transcytosed across the endothelial barrier. With this approach, the EGF is monobiotinylated using an extended polyethyleneglycol (PEG) linker, in parallel with the conjugation of streptavidin (SA) to the TfR MAb (43) . Owing to the very high affinity of SA binding of biotin, there is immediate capture of the EGF–PEG–biotin by the TfR MAb–SA conjugate. The attachment of the EGF to the TfR MAb results in the formation of a bifunctional molecule, called a chimeric peptide, that both binds the BBB TfR to enable entry into the brain from blood, and to the EGF receptor (EGFR) on the cancer cell to enable neuroimaging. In addition, the EGF is conjugated with a diethylenetriaminepentaacetic acid (DTPA) moiety to allow for chelation of the 111 Indium radionuclide. When the unconjugated [111 In]-EGF is injected intravenously into tumor-bearing rats, there is no imaging of a large brain cancer because the EGF peptide radiopharmaceutical does not cross the BBB even in the vicinity of the cancer (Figure 5⇓. However, when the [111 In]-EGF chimeric peptide is administered intravenously, there is imaging of the brain cancer expressing the EGFR (Figure 5B⇓, panels 1 and 2). This model could be replicated for hundreds of endogenous neuropeptides to allow for imaging in vivo of peptidergic neurotransmission systems within the brain. However, peptides cannot be used as radiopharmaceuticals or as new diagnostic agents for the brain unless they are reformulated to enable BBB transport.
Delivery of protein therapeutics to the brain. A. Structure of an epidermal growth factor (EGF) chimeric peptide formed by conjugating the EGF to a molecular Trojan horse consisting of a monoclonal antibody (MAb) to the BBB transferrin receptor (TfR). See text for details. Thus, the EGF chimeric peptide is a bifunctional molecule that binds to the BBB TfR to allow for transport across the BBB, and to the EGF receptor (EGF-R) to allow for sequestration on the brain cancer cell membrane. Reprinted with permission (42). B. Panels 1 and 3 are autopsy sections of nude-rat brain bearing human U87 gliomas and stained with a MAb that binds the human EGF-R. The size of the tumor is visualized with the immunocytochemistry. Panels 2 and 4 are brain scans of the same rats as shown in Panels 1 and 3, but prior to sacrifice. The live nude rats bearing intracranial U87 human gliomas were administered intravenously either [111In]-EGF alone (Panel 4) or the [111In]-EGF-MAb chimeric peptide (Panel 2), indicating that EGF alone does not cross the BBB (Panel 4). However, the tumor is visualized with EGF chimeric peptide owing to transport of the EGF chimeric peptide across the BBB in the tumor (Panel 2). Reprinted with permission (42). C. Structure of a chimeric peptide of brain derived neurotrophic factor (BDNF) that is conjugated to a TfR MAb through an SA–biotin (B) linkage. The BDNF chimeric peptide is a bifunctional molecule that can bind both the BBB TfR to allow for transport from blood to brain, and the neuronal trkB receptor to allow for neuroprotection in brain. Reprinted with permission (47). D. Coronal sections of rat brain stained with 2,3,5-triphenyltetrazolium chloride (TTC). Coronal sections are shown for four different rats in four different treatment groups including saline, BDNF alone, TfRMAb alone, or the BDNF–TfR MAb chimeric peptide. The BDNF was administered at a dose of 50 μg/rat intravenously following permanent occlusion of the middle cerebral artery. The coronal slabs were scanned and the grayscale image was inverted and colorized so that the infarcted region appears dark purple and the healthy brain tissue appears yellow/red. There is no reduction in stroke volume with the BDNF alone because the neurotrophin does not cross the BBB, even in the infarcted region of brain. However, there was a 65% reduction in stroke volume following the delayed intravenous injection of the BDNF chimeric peptide because the neurotrophin was enabled to cross the BBB and enter into the ischemic brain region. Reprinted with permission (47).
Recombinant Proteins as Neurotherapeutics
Many neurotrophins are neuroprotective when these proteins are injected directly into the brain prior to brain ischemia or injury (44) . The neurotrophins must be injected into the brain because these large molecules do not cross the BBB. Therefore, in the absence of BBB disruption, neuroprotection is not possible following delayed intravenous administration of the neurotrophin. Presently there is no neuroprotective agent in clinical practice available for patients with acute stroke or injury (45) . Neuroprotectives have failed in clinical trials of stroke because the drugs are either too toxic or do not cross the BBB. Although the BBB becomes disrupted in later stages of a stroke when neuronal survival is no longer possible, the BBB is intact in the first few hours after stroke when death of ischemic neurons can still be prevented. Recombinant neurotrophins such as brain-derived neurotrophic factor (BDNF) can be used for neuroprotection following delayed intravenous administration in stroke if the neurotrophin is reformulated to enable transport across the BBB. The structure of a BDNF chimeric peptide following conjugation to BBB transport vector is shown in Figure 5C⇑. The biologic activity of the BDNF chimeric peptide was tested in both global and regional brain ischemia models (46–48) . In global brain ischemia, there was complete neuroprotection of the pyramidal neurons of the CA1 sector of the hippocampus seven days after transient forebrain ischemia (TFI). In contrast, intravenous administration of the BDNF alone did not cause any neuroprotection in the TFI model (46) because BDNF does not cross the BBB and the BBB is intact in the early phases after global brain ischemia. Regional brain ischemia is induced with the middle cerebral artery occlusion (MCAO) model. There is no reduction of stroke volume following the intravenous administration of BDNF in either permanent (47) or reversible (48) MCAO (Figure 5D⇑). However, there is a 65–70% reduction in stroke volume when BDNF is conjugated to a molecular Trojan horse and administered intravenously as a chimeric peptide (Figure 5D⇑). There are many other recombinant proteins that could enter CNS drug development pathways if these proteins were reformulated to enable BBB transport. The reformulation of a protein therapeutic to enable BBB transport can be accomplished with genetic engineering and the construction of fusion proteins of the BBB transport vector and the protein therapeutic (2) . Alternatively, fusion proteins can be genetically engineered that comprise the transport vector and avidin, and the vector/avidin fusion protein can be combined with mono-biotinylated protein or antisense therapeutic (2) . The re-formulation of a large-molecule drug to enable transport across the BBB is technically simpler than the laborious and uncertain process of attempting to discover a small-molecule peptidomimetic. Moreover, if the Mr of the peptidomimetic is greater than 400–500 Da, or if the molecule is water soluble, the small-molecule peptidomimetic will still have to be reformulated to enable BBB transport.
Blood-Brain Barrier Transport of Nonviral Gene Medicines
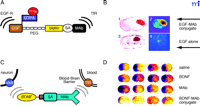
To date, no diseases of the brain have been treated effectively with gene therapy because the viral vectors that are used in gene therapy do not cross the BBB. The intracerebral implantation of the viral vector may provide some therapeutic effect in the rodent brain, but the craniotomy approach is generally not feasible in the human brain, which is approximately 1000-fold larger than the brain of a rat or mouse. Even in the rat brain, an intracerebral implant only distributes drug or gene therapy to a small volume at the tip of the injection needle or border of the implant (Figure 2A⇑). There are also concerns about the long-term effects of the permanent and random alteration of the host genome by viruses such as retrovirus or adeno-associated virus. In contrast to craniotomy, the vascular route to brain (Figure 2C⇑) does enable the global expression of a therapeutic gene throughout the brain. However, gene delivery to brain across the vascular barrier requires the use of BBB gene-targeting technology and molecular Trojan horses. In this approach, a nonviral supercoiled plasmid DNA is encapsulated in the interior of an 85 nm liposome (49) . Any DNA located on the outside of the liposome is exhaustively removed by nuclease treatment. The surface of the liposome is conjugated with 1000–2000 strands of 2000-Da PEG to form a “pegylated” liposome. DNA encapsulated in pegylated liposomes is stable in blood and has a prolonged blood residence time (50) . However, the pegylated liposome is relatively inert and is not taken up by brain. Therefore, the tips of 1–2% of the PEG strands are conjugated with a peptidomimetic MAb. The conjugation of this molecular Trojan horse to the pegylated liposome forms a pegylated immunoliposome (PIL). The relationship of the targeting ligand to the liposome is visualized with electron microscopy (Figure 6A⇓) (51) . The targeting MAb enables the PIL carrying the plasmid DNA to bind to cell surface receptors, as shown in Figure 6B⇓, followed by receptor-mediated transcytosis across the BBB and receptor-mediated endocytosis across the neuronal cell membrane of the PIL.
Non-invasive, non-viral gene transfer to the primate brain. A. Transmission electron micrograph of a pegylated immunoliposome (PIL). The IgG molecules tethered to the tips of the 2000-Da polyethyleneglycol (PEG) are bound by a conjugate of the 10 nm gold and an mouse-specific secondary antibody. The position of the gold particles illustrates the relationship of the PEG-extended monoclonal antibody (MAb) and the liposome. Magnification bar = 20 nm. Reprinted with permission (51). B. Plasmid DNA encapsulated in the interior of the PIL, which is conjugated with a receptor (R)-specific targeting MAb. The targeting MAb is conjugated to the tips of 1–2% of the PEG strands that project from the surface of liposome, and there are about 2000 strands of PEG conjugated to the liposome surface. The PEG strands inhibit uptake of the PIL by the reticulo-endothelial system in vivo and enable a prolonged blood residence time of the PIL in vivo (50). The tissue-specific expression of the exogenous gene in vivo can be regulated with the use of tissue-specific promoters incorporated into the plasmid DNA (53). C. Tyrosine hydroxylase (TH) immunocytochemistry of rat brain 3 days after a single intravenous injection of a TH expression plasmid encapsulated in a PIL and targeted to neurons of brain with either a MAb to the BBB TfR (left panel) or a mouse IgG2a isotype control antibody (right panel). Adult rats received an injection of the neurotoxin, 6-hydroxydopamine, on the right side of the brain into the median forebrain bundle 4–5 weeks prior to the intravenous gene therapy. The successful creation of the neurotoxin lesion was confirmed by testing rotation behavior prior to TH gene therapy (51). There is complete normalization of both striatal TH expression and activity ipsilateral to the neurotoxin injection when the TH expression plasmid is effectively delivered to the brain with a TfR Mab–targeted PIL (left panel), because the PIL is able to traverse the BBB via transport on the TfR. However, there is no reconstitution of striatal TH with the control PIL (right panel), because this PIL is unable to cross the BBB. Reprinted with permission (51). D. β-galactosidase histochemistry of brain removed from an adult Rhesus monkey 48 hours after a single intravenous injection of a β-galactosidase-expressing plasmid encapsulated in a PIL conjugated to a human insulin receptor (HIR)-specific MAb. There is global expression of the exogenous gene throughout the primate brain with increased expression in gray matter relative to that in white matter. Panels (E) and (F) are light micrographs of occipital cortex and cerebellum, respectively. The columnar organization of the occipital cortex of the primate brain is revealed (E), and the dense gene expression in the molecular and granular layers of the cerebellum and the intermediate Purkinje cells, are visible in (F). Panels D–F reprinted with permission (49).
Gene Therapy of Brain Cancer
Human U87 glioma cells injected into the brain of severe combined immunodeficiency (SCID) mice leads to the development of intra-cranial brain cancer (52). The human cancer was perfused by blood vessels of mouse brain origin. In order to deliver a therapeutic gene to this cancer, it was necessary to traverse two barriers in series: the mouse BBB, and the human tumor–cell membrane. For gene delivery across the mouse BBB, a rat MAb (8D3) that binds to the mouse TfR is used (53) . Gene delivery to human cells is accomplished with a murine MAb (83-14) that recognizes the human insulin receptor (HIR). Thus, the PIL was doubly conjugated with both the 8D3 and 83-13 Mabs (52). With this system, gene therapy of brain cancer was possible with an intravenous injection of a nonviral formulation. The delivery of a gene encoding antisense RNA to the human EGFR caused a 100% increase in survival time—twice as long as those tumor-bearing mice receiving PIL expressing a control gene (luciferase) (52).
Gene Therapy of Experimental Parkinson Disease
One animal model of PD involves the injection of the neurotoxin 6-hydroxydopamine into the medial forebrain bundle of rats. This toxin disrupts the dopaminergic pathway between the substantia nigra and the striatum, and the subsequent expression of striatal tyrosine hydroxylase (TH) is almost completely blocked ipsilateral to the toxin injection. A nonviral expression plasmid that encoded rat TH was encapsulated in PILs and targeted to rat brain by a murine MAb (OX26) that binds to the rat TfR (50) . Owing to the presense of the TfR on both the BBB and the neuronal cell membrane, the OX26-targeted PIL carrying the TH gene was delivered across both the BBB and the neuronal plasma membrane. With this approach, intravenous nonviral gene therapy caused a 100% normalization of striatal TH activity in the 6-hydroxydopamine-lesioned rat (Figure 6C⇑) (51).
Global Gene Delivery to the Primate Brain
Gene delivery to the brain of primates or humans is possible with a peptidomimetic MAb specific for the HIR (49). The HIR MAb is a highly active transport vector, and the level of expression of an exogenous gene, luciferase, in the primate brain targeted with the HIR MAb is 50-fold higher than the level of luciferase gene expression in rat brain targeted with a TfR MAb (49) . The delivery of a β -galactosidase expression plasmid across the BBB following the intravenous injection in the rhesus monkey is shown in Figure 6D⇑. Virtually every neuron of the brain expresses the exogenous gene because the plasmid DNA was delivered to brain via transvascular route (Figure 2C⇑). The neurons of the cortical columns of the occipital cortex (Figure 6E⇑) or of the cerebellar cortex (Figure 6F⇑) of the primate brain express the exogenous gene. Pharmacological effects of gene therapy delivered with the PIL gene targeting technology are possible because there is such a high rate of gene transfection of brain cells with this approach. The normalization of striatal TH activity was possible with the delivery of only five to ten plasmid DNA molecules per brain cell (51) . Each plasmid may then produce many copies of the expressed mRNA, which in turn produces many copies of the protein.
Gene Therapy of the Human Brain
The molecular Trojan horse antibodies projecting from the surface of the PIL are visualized by electron microscopy as shown in Figure 6A⇑. The only immunogenic component of this formulation is the MAb, and the immunogenicity of the Trojan horse in humans can be reduced or eliminated with genetic engineering and the production of a “humanized” MAb. (Following the genetic engineering, the amino acid sequence of a humanized MAb is 95% human sequence and 5% mouse sequence.) A genetically engineered chimeric form of the HIR MAb has been produced and has the same avidity for the HIR in vitro or at the primate BBB in vivo, as the original murine HIR MAb (54) . Therefore, the technology is now available for the noninvasive delivery of nonviral gene medicines to the human brain, making it feasible to create adult transgenic patients within twenty-four hours of delivering the therapy.
Blood-Brain Barrier Genomics
The outline in Figure 3⇑ emphasizes the many pathways available for the development of effective BBB drug delivery strategies with a biology-based approach that focuses on endogenous BBB transport systems. The future discovery of CMT, AET, or RMT systems at the BBB can be accelerated with the development of a BBB genomics program (35, 55, 56). A successful BBB genomics program would necessarily be separate from a brain genomics program because the volume of the capillary endothelium in brain is < 10-3 of the brain’s total volume (2) and the sensitivity of most gene microarrays is on the order of 10-4 (57) . Thus, screening a whole-brain gene microarray would surely miss many BBB-specific transcripts. A BBB genomics program starts with the initial isolation of animal or human brain microvessels (Figure 7⇓), which constitute approximately 0.1% of the whole brain volume. From these microvessels, the BBB specific mRNA is subsequently isolated for production of BBB specific cDNA. A BBB genomics program for either animal or human brain has been developed using the subtractive suppressive hybridization (SSH) methodology (58) for selecting BBB-enriched genes. In this approach, cDNA derived from brain capillary RNA is used to prepare a “tester” cDNA library. In parallel, a “driver” cDNA library is produced from RNA pooled from liver or kidney or any alternative source of RNA. The driver cDNAs will be matched up to specific tester libraries to remove (subtract) non-specific cDNAs. The subtracted tester cDNA library is then screened with subtracted tester cDNA. The initial application of the BBB genomics methodology has led to the discovery of nearly 100 BBB specific gene products (35, 55, 56). These genes fall into two categories (Figure 7⇓). About half of the genes discovered in the BBB genomics program are known genes that are selectively expressed at the BBB (Figure 7⇓). The other half of the genes discovered are either found in the expressed sequence tag (EST) database or are new and uncharacterized genes not found in any nonhuman database. These considerations suggest that no more than half of the functional BBB transporters have been discovered to date. The future discovery of novel BBB transporters can be accelerated with a BBB genomics program.
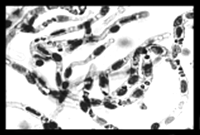
Outline of a BBB genomics program. Starting with the isolation of brain capillaries (above) from either fresh human brain or animal brain, libraries of brain capillary cDNAs are produced following the initial isolation of brain capillary derived polyA+ RNA. Screening for BBB-enriched genes using methodology such as suppressive subtractive hybridization (SSH) leads to classification of genes based on whether the gene function is known or unknown. The genes of unknown function represent about 50% of the detected genes (35, 55, 56). Genes of unknown function consist of uncharacterized genes and gene fragments found in expressed sequence tag (EST) databases. Genes of known function can be categorized into a variety of different gene families as outlined in (35, 55, 56). These genes of known function are selectively enriched at either the rat or human BBB; the names of these known genes are given in full in the original publications (35, 55, 56).
Our growing knowledge of the BBB endogenous transporters will provide the platform for the development of future BBB drug delivery strategies. The merging of CNS drug-targeting with CNS drug-discovery can address the present-day challenges in CNS drug-development. Solutions to the BBB problem can lead to the treatment of many CNS disorders that may not be treatable with current models of CNS drug development that rely solely on lipid soluble small molecules. If BBB drug targeting is not incorporated into CNS drug discovery, then future innovations in CNS drug development will be limited to lipid soluble small molecules, which largely treat only a few diseases such as depression, schizophrenia, chronic pain, and epilepsy.
Conclusions
The incorporation of BBB drug delivery strategies within the global CNS drug-development effort is virtually nonexistent. Considering the rate-limiting role played by the BBB in the development of nearly all new drugs for the brain, it is difficult to understand why the BBB has been so consistently underdeveloped in both academic and industry laboratories. Even if a pharmaceutical company wanted to reverse this trend, it would be difficult to hire a critical mass of scientists trained in the BBB. This is because there are no academic programs that specialize in BBB transport biology within Departments of Neuroscience or Departments of Pharmacology in the United States. However, a few Departments of Pharmaceutical Chemistry within Schools of Pharmacy are now building BBB transport biology programs. Given the chronic underdevelopment of BBB transport biology within academic neurosciences, there is no worldwide infrastructure or critical mass of scientists trained in BBB transport biology. This lack of global BBB infrastructure is the single most important factor that will limit the future of brain drug development.
Acknowledgments
This work was supported by the NIH and the US Department of Energy.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

William M. Pardridge, MD , is Professor of Medicine at the UCLA School of Medicine. E-mail wpardridge{at}mednet.ucla.edu;
fax 310-206-5163.

