LTP May Trigger Addiction
Widely used classes of abused drugs have very different chemical structures and, therefore, different initial molecular targets in the brain. Nevertheless, there has been great interest in identifying a common substrate for their actions. The hope is that this will uncover a target for therapeutic intervention in addiction. Two broad similarities between drug classes have emerged. First, drugs of abuse share with natural rewards, such as food and sex, the ability to increase dopamine transmission in the mesocorticolimbic dopamine system (Figure 1⇓). Dopamine cell bodies of this system originate in the ventral tegmental area (VTA) and project to cortical and limbic regions. Of these, the nucleus accumbens is considered most critical for motivated behavior, as it serves as an interface between limbic and motor systems (1) . A second common feature, recognized more recently, is that drugs of abuse influence neuronal plasticity in brain pathways related to motivation and reward (2) . Long-term potentiation (LTP) is the leading candidate for mediating neuronal plasticity and has therefore become a focus of addiction research. The most intensely studied form of hippocampal LTP requires N -methyl-d -aspartate (NMDA) receptor stimulation for its development, but is expressed as an enhancement of α -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor transmission as a result of AMPA receptor insertion into postsynaptic sites (3) .
In the February 2003 issue of Neuron, Saal et al. (4) linked these two areas of addiction research by demonstrating that five different drugs of abuse (i.e., amphetamine, cocaine, morphine, ethanol, and nicotine) or a stressful episode (swimming in cold water) produce LTP at excitatory synapses onto VTA dopamine neurons. To understand the implications of this finding, it helps to consider how plasticity came to be associated with drug addiction.
The idea originated, in large part, from studies of behavioral sensitization—an animal model for addiction (5 , 6) . Sensitization refers to the intensification of behavioral responses to drugs of abuse that occurs when they are administered repeatedly. Sensitization of locomotor stimulatory effects is commonly studied, but more importantly, sensitization develops to the incentive–motivational properties of drugs, that is, the properties that make them “wanted” (6) . Thus, an amphetamine preexposed rat will work harder to obtain the drug than a naïve rat, and an addict will continue to seek and take drugs despite enormous adverse consequences (7) . Animals remain sensitized for months to years, reminiscent of the persistence of vulnerability to relapse in recovering addicts; therefore, it is notable that many drugs of abuse produce sensitization and that there are many examples of cross-sensitization between different drug classes.
Repeated injections of amphetamine or morphine into the VTA are sufficient to produce sensitization whereas a challenge injection directly into the nucleus accumbens (but not VTA) will elicit a sensitized locomotor response in rats previously treated with repeated systemic or intra-VTA drug injections. Anatomical separation of sites for initiation and expression implies “transfer” of sensitization from VTA to forebrain regions such as nucleus accumbens. Many lines of evidence suggest that the “transfer” mechanism involves a transient increase in the activity of VTA dopamine neurons during the first several days after discontinuing drug administration. Conversely, treatments that transiently activate dopamine neurons, such as electrical stimulation of the VTA, will result in an animal that appears sensitized to drugs (5) .
A breakthrough came from the demonstration that behavioral sensitization, like LTP, requires glutamate transmission (8) . Later studies pinpointed the VTA as a necessary site for glutamate transmission, leading to the suggestion that the initiation of sensitization involved LTP at excitatory synapses onto VTA dopamine neurons (5) . This hypothesis would account for the increase in dopaminergic cell activity that is critical for the early stages of sensitization. Indirect evidence supporting this hypothesis—provided by electrophysiological and microdialysis studies—showed that dopaminergic neurons, during early withdrawal from cocaine or amphetamine, are more sensitive to the excitatory effects of AMPA (9 , 10) .
However, it was not until 2001 that behavioral sensitization was directly linked with the induction of LTP in VTA dopamine neurons. Malenka, Bonci, and colleagues compared excitatory transmission in midbrain slices from control mice and mice injected the day before with a single injection of cocaine (11) . The dose of cocaine used produced so-called “one-shot” behavioral sensitization. In slices from cocaine-treated mice, they observed an increase in the portion of the excitatory postsynaptic current mediated by AMPA receptors relative to NMDA receptors (the AMPA/NMDA ratio). An increase in this ratio is diagnostic for LTP, reflecting insertion of new AMPA receptors into postsynaptic sites (3) . Cocaine-induced LTP was correlated with cocaine-induced behavioral sensitization, as both were prevented if cocaine was coadministered with an NMDA receptor antagonist. Moreover, further LTP could not be elicited in dopamine neurons from sensitized rats, consistent with the idea that LTP was already maximally induced by cocaine.
In Saal et al. (4) , the same group extended their findings by asking if different classes of abused drugs share the ability to produce LTP in VTA dopamine neurons, again using the AMPA/NMDA ratio as a marker for LTP. They also examined the effect of a stressful episode. In recovering addicts, stress is a common trigger for relapse. In rats, there is cross-sensitization between the locomotor response to stress and drugs, and stress enhances vulnerability to acquire and maintain drug self-administration (12) . Saal et al. found that the AMPA/NMDA ratio was increased in brain slices prepared one day after a single injection of amphetamine, cocaine, morphine, ethanol, or nicotine. An even greater increase was produced by a stressful episode. Whereas an NMDA receptor antagonist prevented the response to both cocaine (11) and stress (4) , consistent with the idea that both produce NMDA receptor–dependent LTP, an antagonist of glucocorticoid receptors prevented the response to stress but not cocaine (4) . Thus, the synaptic change produced by cocaine is not attributable to stress associated with the drug’s actions.
Perhaps the most fundamental question raised by these findings is how different drugs produce LTP. LTP is elicited by high frequency activation of a glutamate synapse. But, of the five drugs tested, only nicotine directly augments glutamate release and LTP in the VTA (13) . The other drugs activate dopamine neurons indirectly, often by removal of inhibitory tone through actions in the VTA as well as other brain regions (Figure 1⇓⇓). In fact, the immediate effect of cocaine and amphetamine in the VTA is to inhibit dopamine neurons by increasing extracellular levels of dopamine, which then activates dopamine autoreceptors. A second question is related to the fact that the VTA is the origin of a number of distinct dopamine projections that innervate different cortical and limbic regions and are selectively regulated by afferent inputs (14) . Do all excitatory synapses in the VTA undergo LTP? If not, do all drugs affect the same synapses? Does LTP produced by one drug occlude LTP induction by another?
The questions become more complex when evaluating the significance of the data in the context of behavioral sensitization. Sensitization requires a cascade of events involving different brain regions, neurotransmitters, and signal transduction systems at different stages of its development and maintenance (5) . LTP may contribute to the activation of dopamine neurons during early withdrawal that seems essential for more persistent downstream changes. But its role must be understood in the context of other changes occurring in VTA during early withdrawal, including altered sensitivity of DA autoreceptors and other regulatory mechanisms (15) , enhanced GABAB receptor transmission (16) , and complex changes in neurotrophic factor signaling (17) . Adaptations in other brain regions must also be considered. Although the VTA is critical for initiation of sensitization by psychomotor stimulants and opioids, the anatomy of sensitization to other drugs is poorly understood. Even for stimulants, the development of sensitization is a circuit phenomenon, as sensitization is prevented by lesions of basolateral amygdala and prefrontal cortex, and by glutamate antagonist injections into several brain regions (5) . This cannot be explained simply by positing that these regions provide glutamate afferents to VTA necessary for LTP. For example, prefrontal cortex lesions prevent sensitized responses involving VTA dopamine neurons projecting to the nucleus accumbens (18) , but the prefrontal cortex does not send glutamate projections to the cell bodies of these neurons (14) .
Another important issue is that LTP was demonstrated after one exposure to each drug (4) and only in the case of cocaine was it shown that this single exposure produced locomotor sensitization (11) . Even if all drugs had been shown to produce “one-shot” sensitization, this cannot necessarily be equated with the more commonly studied form of behavioral sensitization produced by repeated drug administration. One major difference is that the expression of “one-shot” sensitization is completely under conditioned control; that is, rats express a sensitized response only when tested in the same environment in which drug was first administered (19) . More importantly, studies demonstrating sensitization of incentive-motivational effects have used repeated drug administration, not the “one-shot” paradigm (7) . Thus, a critical next step is to evaluate the relationship between LTP and sensitization of incentive-motivational effects.
Beyond sensitization, the broader implications of this study for addiction hinge on the enormous question of what dopamine “does” in the reward circuitry. Originally, dopamine was thought to mediate the pleasure evoked by rewarding stimuli, but it was later demonstrated that rats with complete depletions of striatal and nucleus accumbens dopamine are still able to make judgments about whether they “like” or “don’t like” a particular taste (6) . Several more complex theories have evolved. One holds that dopamine neurons “learn and predict” the occurrence of rewards and thus participate in the ongoing learning of adaptive behaviors (20) . This fits well with the ability of dopamine (and drugs of abuse) to facilitate stimulus–reward learning and its impact on behavior (21) . Another influential hypothesis is that dopamine neurons attribute incentive salience to rewards and to conditioned stimuli associated with rewards, thereby enhancing the extent to which they are “wanted” and can therefore shape behavior. Repeated administration of drugs of abuse is proposed to sensitize the dopamine-dependent systems that mediate incentive salience, leading to pathological intensification of drug “wanting” (6). A related hypothesis emphasizes the possible importance of sensitization-related changes in dopamine systems in facilitating incentive learning of drug-related stimuli (22) . All of these theories help explain how chronic drug exposure facilitates the formation of new habits that center around drug-seeking behavior, usually at the expense of more appropriate behaviors. If LTP in the VTA enhances dopamine transmission in cortical and limbic regions essential for motivated behaviors, even for only a few days after discontinuing drug exposure, cues associated with drugs may be attributed greater motivational significance, rendering an individual more susceptible to the “learning” of behaviors that are associated with drug use.
It will be fascinating to work out the mechanisms downstream of LTP-induced dopamine cell activation. We know that dopamine itself and drugs of abuse can influence the basic cell biological mechanisms that enable plasticity (2 , 23) , the pathways that govern gene expression in learning and addiction (24) , and the morphological changes that may mediate long-lasting rewiring of neuronal circuits (6) . The challenge is to determine which of the myriad of drug-induced adaptations actually underlie intensification of drug craving (25) and then devise strategies to counteract them.
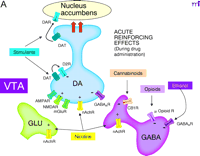
Interactions of reinforcing drugs with the mesoaccumbens dopamine (DA) system, focusing on the ventral tegmental area (VTA). A. Acutely, all reinforcing drugs increase DA transmission in this system. Stimulants (e.g., amphetamine, cocaine) interact with the DA transporter (DAT) to elevate extracellular DA levels, dramatically enhancing DA transmission in nucleus accumbens [but inhibiting DA cell firing through autoreceptors (D2R) in the VTA]. Nicotine, through nicotinic acetylcholine receptors (nAchR) excites DA cells directly and promotes glutamate release from glutamate nerve terminals (GLU) in the VTA. Opioids, through opioid receptors, inhibit -amino butyric acid (GABA) neurons in the VTA, disinhibiting DA neurons. Actions in the nucleus accumbens are also important for opioid reinforcement. Ethanol and cannabinoids also interact with VTA GABA neurons to disinhibit DA neurons. Ethanol is a positive allosteric modulator of GABA A receptors, whereas cannabinoids activate presynaptic CB1 receptors that inhibit GABA release.
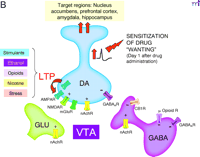
B. Saal et al. (4) showed that on day one after drug administration, five drugs of abuse (i.e., amphetamine, cocaine, nicotine, ethanol, or morphine) or a stressor produced LTP at excitatory synapses onto VTA DA neurons. By transiently activating DA transmission to nucleus accumbens and other forebrain areas, this may trigger sensitization of drug “wanting.”
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Marina E. Wolf, PhD , is Professor and Acting Chair in the Department of Neuroscience at the Chicago Medical School. E-mail marina.wolf{at}finchcms.edu; fax (847)-578-8515.

