S-Nitrosylation Signaling in Cell Biology
Abstract
S-Nitrosylated proteins form when a cysteine thiol reacts with nitric oxide (NO) in the presence of an electron acceptor to form an S-NO bond. Under physiological conditions, this posttranslational modification affects the function a wide array of cell proteins, ranging from ion channels to nuclear regulatory proteins. Recent evidence suggests that 1) S-nitrosylated proteins can be synthesized by exposure of specific redox-active motifs to NO, through transnitrosation/transfer reactions, or through metalloprotein-catalyzed reactions; 2) S-nitrosothiols can be sequestered in membranes, lipophilic protein folds, or in vesicles to preserve their activity; and 3) S-nitrosothiols can be degraded by a number of enzymes systems. These recent insights regarding the bioactivities, molecular signaling pathways, and metabolism of endogenous S-nitrosothiols have suggested several new therapies for disease ranging from cystic fibrosis to pulmonary hypertension.
Introduction
Endogenous S- nitrosothiols (SNOs) are naturally occurring moieties on proteins in which a sulfur atom from cysteine or homocysteine reacts with nitric oxide (NO) to form an S- NO bond. Conventionally, this reaction occurs as an electrophilic attack of a nitrosonium (NO+ ) equivalent on sulfur, followed by depronation. The convention involves attack of nitrosonium on the thiolate anion; however, reactions involving nitroxyl (NO− ) equivalents or NO radicals have also been demonstrated (1, 2) . Within mammalian tissues, the concentration of SNOs can vary from nM to μ M levels (1, 3, 4) , and thiol S- nitrosylation and NO transfer reactions (transnitrosation reactions) are involved in virtually all classes of cell signaling, ranging from regulation of ion channels and G-protein coupled reactions to receptor stimulation and activation of nuclear regulatory proteins. Furthermore, it is now apparent that the synthesis, transport, activation, and catabolism of SNOs are regulated.
Biosynthesis of Endogenous SNOs
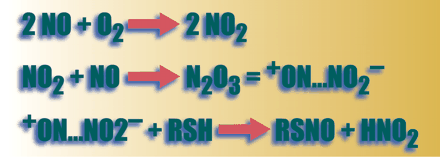
Synthesis of S- nitrosothiols from NO in biological systems generally requires the presence of an electron acceptor. It was once assumed that this electron acceptor must be oxygen, according to the third order reaction (a)
-
2 NO + O2 → 2 NO2 followed by,
-
NO2 + NO → N2 O3 = + ON…NO2−
-
+ ON…NO2− + RSH → RSNO + HNO2 (where R is the substrate to be nitrosylated)
The rate-limiting reaction (a) proceeds relatively slowly under physiological conditions, where NO concentrations are nanomolar and oxygen concentrations are micromolar, though rate constants in lipid phase and in lipophilic protein pockets (8.8 x 107 M— 2.S— 1) are substantially greater than those in aqueous phase (6.6 x 106 M— 2.S— 1) (5–8) . Additional inorganic electron acceptors, such as NAD+ , can facilitate SNO formation (9) , and there is also evidence that iron–nitrosyl species might catalyze SNO formation in vivo; in the latter case, the iron species participates as the electron acceptor (10) .
More recently, it has been appreciated that specific proteins catalyze SNO synthesis. Indeed, there are consensus motifs consisting of a core of three residues, K/R/H/D/E–C–D/E, that predict S- nitrosylation of cysteines in hydrophilic protein domains (11) . For instance, arginine and aspartate residues that flank Cys131 in methionine adenosyl transferase serve as partial electron acceptors, making NO more electrophilic and reactive with the target thioate (12) .
Ceruloplasmin serves as a model enzyme for the synthesis of low molecular weight SNOs, particularly S- nitrosoglutathione (GSNO) (13) . Type I copper (Cu+ ) serves as the electron acceptor whereby the electron is shuttled from Cu+ to additional coppers in ceruloplasmin, and the NO+ transferred to the thiolate on glutathione, for example. There is a net four-electron oxidation of O2. Hemoglobin also functions as a redox-sensitive metalloprotein that can catalyze SNO formation (14, 15) . NO can react with deoxy (T-state)-ferrous hemoglobin to form a heme iron-nitrosyl intermediate. Upon oxygenation, a shift in conformational equilibrium occurs converting hemoglobin from T → R state, whereby the nitroso group intramolecularly transfers to Cysβ93 . The nitrosothiol bond at Cysβ93 , however, is sterically hindered on return to the T state. With the next cycle of deoxygenation, the NO moiety, not the NO+ moiety, returns to the heme, completing the intramolecular Fe ↔ S shuttle, or is offloaded for erythrocytic export perhaps by transnitrosation of the anion exchange protein 1 (AE1) or low molecular weight thiols such as glutathione (14–17) .
S- Nitrosothiol synthesis does not always require electrophilic attack of NO+ on thiolate. There is evidence that nitroxyl equivalents (NO− ) may attack relatively electropositive cysteine-sulfur groups, as in the case of S- nitrosylation of the N -methyl-d -aspartate (NMDA) receptor (18, 19) . SNOs are also formed following the activation of any of the three nitric oxide synthase (NOS) isoforms and can occur directly through NO− formation by NOS or through coproduction of NO and superoxide by NOS in the presence of glutathione (20) . More recently, Gow and coworkers have demonstrated, using SNO-specific antibodies, the direct formation of SNOs following acetyl choline–induced NOS 3 activation, NOS 1 activation, and cytokine-induced NOS 2 activation in a variety of cell types and organ preparations (21) .
In summary, although inorganic chemical reactions involving N2 O3 and other intermediates may be relevant to physiological SNO formation in biological membranes and hydrophobic pockets of proteins, protein-mediated or -catalyzed formation is increasingly appreciated as an important determinant of SNO formation in many cell systems. These latter biochemical processes can involve: 1) activation of NOS isoforms themselves, 2) reactivity of NOS- derived NO− /NO/NO+ equivalents with target protein consensus motifs, and 3) metalloprotein-catalyzed reactions such as those carried out by ceruloplasmin. The spectrum of SNO synthetic reactions may be analogous to kinase reactions in phosphorylation signaling but are substantially less well understood.
S-Nitrosothiol Catabolism
A number of enzyme systems catabolize S- nitrosothiols in vitro. These include xanthine/xanthine oxidase (X/XO) (22) , thiodoxin/thioredoxin reductase (T/TR) (23, 24) , γ glutamyl transpeptidase (γ GT) (17, 25) , glutathione peroxidase (26) , copper zinc superoxide dismutase (Cu/Zn SOD) (27, 28) and glutathione-dependent formaldehyde dehydrogenase (GDFDH, which has been referred to as GSNO reductase) (29, 30) . There is compelling evidence that Cu/Zn SOD, T/TR, γ GT, and GDFDH participate in the normal regulation of cellular SNO levels. Discussion of these enzymes will be prefaced by the introduction of two background concepts: transnitrosation reactions and S- nitrosothiol compartmentalization.
Transnitrosation Reactions
Transnitrosation is the process by which an NO equivalent is transferred from one molecule to another. Transfers between thiol groups are vastly favored over transfers to nitrogen- or carbon-containing species (31) . Equilibria exist between low-mass and protein SNOs in cellular and interstitial SNO pools (32, 33) , but although on- and off-rate constants for these equilibria in vitro have been measured (33, 34) , these kinetics may be less relevant in vivo than enzymatic processes. For example, simple inorganic transnitrosation equilibria cannot explain the fact that GSNO concentrations are on the order of 7 μ M in the rat medulla (4) and are undetectable in the thalamus (34) , whereas S- nitrosocysteinyl glycine is the principal low-mass S- nitrosothiol in the thalamus (34) . These concentrations appear to be regulated by specific GSNO catabolic enzymes.
Compartmentalization of S-Nitrosothiols
The cytosol is perhaps the least conducive cellular environment for SNO stability because protein SNOs can be readily reduced by glutathione or thioredoxin, each of which, once S- nitrosylated through transnitrosation, can be enzymatically denitrosylated (24, 26, 30) . To protect S- nitrosothiols from reductive or transnitrosative degradation, they may be stored or protected in membranes, in lipophilic protein folds, in vesicles, and in interstitial spaces (7, 35, 36) . Caspase activation during apoptosis provides an example of how this type of sequestration is used in the process of cell signaling. These enzymes are ordinarily sequestered in an S- nitrosylated (inactive) state in the mitochondrial intermembrane space. When a cell receives an apoptotic signal such as Fas–Fas ligand binding, these caspases are released into the cytosol where they rapidly become denitrosylated. Denitrosylation leads to enzyme activation and initiation of apoptosis (36, 37) .
Inorganic S-nitrosothiol catabolism
Several inorganic processes can lead to cleavage of the SNO bond (1, 38) . These include photolytic cleavage as well as cleavage caused by reactions with inorganic copper or mercury. However, these inorganic reactions are likely to be of little physiological relevance: free copper and mercury, for example, are nearly undetectable in cell systems (39) . Thus, enzymatic processes appear to be the most important determinant of SNO concentration in cell biology.
Enzymatic S-nitrosothiol catabolism
Glutathione-dependent formaldehyde dehydrogenase [GDFDH, also known as GS-FDH or alcohol dehydrogenase III (ADH III)] is ubiquitously expressed. Indeed, it is much more widely expressed than can be justified by its role as a formaldehyde dehydrogenase. Several studies have shown that GDFDH serves as a GSNO lyase or terminase with a Km in physiological range (∼20 μ M) for eukaryotic cells (29, 30) . It is interesting to note that the enzyme’s catalytic efficiency for GSNO (kcat /Km ), 94,300 mM−1 min−1 , exceeds that of all other ADH III substrates (29) , suggesting that GSNO catabolism could be one of the most important reasons for the ubiquitous expression of ADH III.
Liu and coworkers have created E. coli , yeast, and mice deficient in GDFDH (30) . The wild types of these organisms have low nanomolar concentrations of cytosolic SNOs, almost exclusively represented as SNO–proteins; however, in the absence of GDFDH, cytosolic SNO–protein concentrations are increased, and cytosolic GSNO concentrations become detectable (Figure 1⇓). These authors have proposed that GDFDH serves more to protect against nitrosative stress than as a cell-signaling enzyme because of its ubiquitous nature .
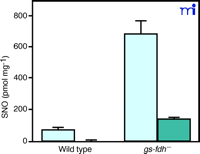
Increased levels of intracellular S-nitrosothiols in gs-fdh— mutant cells after GSNO treatment. Mid-log phase (absorbance 600 nm = 0.4–0.6) cells were cultured in the presence of 5 mM GSNO at 30°C for 2 h. SNO signal in the whole lysate (light blue bars) and the fraction that passed through a 5K cut-off membrane (dark blue bars) were normalized against whole cell lysate protein content. Reprinted with permission (30).
The thioredoxin/thioredoxin reductase system also appears to regulate cytosolic SNO levels, although the actual mechanism remains unknown. Thioredoxin appears to be S- nitrosylated at Cys69 , whereas the redox-active cysteines Cys32 and Cys35 are not S- nitrosylated (24) . S- nitrosylated thioredoxin can, in turn, be reduced by thioredoxin reductase (22) . As with GDFDH, the T/TR system protects the intracellular environment from excessive nitrosative stress. In this sense, localization of thioredoxin reductase activity might serve to regulate SNO trafficking.
Cu/Zn SOD catalyzes the decomposition of low molecular weight nitrosothiols to form NO (27, 28) . GSNO and other S- nitrosothiols are neuroprotective at physiological concentrations (19) . We have recently shown that Cu/Zn SOD mutations associated with familial amyotrophic lateral sclerosis lead to accelerated GSNO catabolism (28) (Figure 2⇓), suggesting that Cu/Zn SOD has an important physiological role in low-mass SNO catabolism, at least in the central nervous system.
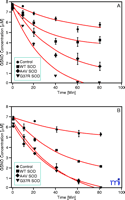
Cu/Zn mutations accelerate the decomposition of GSNO. Decomposition of 7 μM GSNO in the presence of 125 μM glutathione and 10 μM WT SOD (squares), 10 μM A4V SOD (diamonds), 10 μM G37R SOD (triangles) or 0 μM SOD control (circles) in 10 mM PBS at pH 7.4 and 37oC. GSNO content analyzed by ( A) chemiluminenscence or ( B) liquid chromatograpy–mass spectrometry (LC-MS). Reprinted with permission (28).
γ GT catalyzes the decomposition of GSNO to form glutamate and S- nitrosocysteinyl glycine (CGSNO) (25, 40) . GSNO does not readily cross cell membranes, whereas CGSNO does. The bioactivities of physiologically relevant concentrations of GSNO in augmenting effects such as hypoxia inducible factor (HIF-1)-mediated transcriptional activity (41) and in stabilizing in the cystic fibrosis transmembrane regulatory protein (CFTR) (42) are prevented by the γ GT inhibitor, acivicin (Figure 3⇓). Furthermore, GSNO whether produced 1) by NOS 1 in the nucleus tractus solitarius (NTS) as a result of stimulation by afferents from the carotid body or 2) directly by hemoglobin deoxygenation appears to have a critical role in signaling the mammalian ventilatory response to hypoxia (17) . This effect of GSNO injected into the NTS is inhibited completely by acivicin, and the inhibition is overcome by administration of CGSNO (Figure 3⇓). Moreover, animals deficient in γ GT and not supplemented with of N -acetyl cysteine have dramatically abnormal ventilatory recovery from hypoxia. Taken together, these observations suggest that GSNO catabolism by γ GT has an important physiological role in modulating SNO signaling.
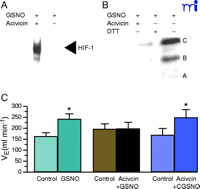
Acivicin reverses the effects of GSNO. A. GSNO-induced HIF-1 DNA binding activity is reversed by acivicin. Nuclear extracts made from bovine pulmonary artery endothelial cells were treated with 100 μM GSNO in the absence (–) or presence (+) or 100 μM acivicin for 4 h. HIF-1 DNA binding activity was determined by electrophoretic mobility shift assay using 3 μg nuclear protein and 1.5 fmol of a 30-bp oligonucleotide containing the HIF-1 DNA binding site. Reprinted with permission from (41) B. GSNO induction of CFTR maturation is inhibited by acivicin and reversed by DTT. Western blot anlysis was performed on 100 μg of wholecell extracts made from CFPAC-1 cells treated with 1 μM GSNO in the presence of 100 μM acivicin for 4 h or 200 μM DTT drug in the last 30 min of the 4 h incubation time. Reprinted with permission (42) C. The GSNO induced-increase in minute ventilation (VE) is blocked by acivicin. VE increases stimulated by microinjection of 10 nmol GSNO were abolished after pretreatment with the gamma glutamyl transpeptidase inhibitor acivicin (7.5 nmol). CGSNO (10 nmol) stimulated VE increased were not modified by acivicin. * indicates statistically significant differences compared to controls. Reprinted with permission (17).
Despite these recent advances, it must be emphasized that specificity in the regulation of SNO catabolism remains to be explained. The pathways described so far primarily involve transnitrosation to low-mass nitrosothiols and subsequent catabolism and/or storage of SNO bioactivity pools in sequestered locations. The story is likely to involve a higher level of complexity.
Bioactivities of S-Nitrosothiols
General Mechanisms of Action
As a general rule, S- nitrosylation reactions cause specific physiological or pathophysiological activities by modifying protein function. Protein activity may be increased (e.g., p21ras or thioredoxin) or inhibited (e.g., caspases, or methionine adenosyl transferase) by S- nitrosylation of specific systems (12, 24, 37, 43) . As reviewed previously, S- nitrosylation of protein thiols may occur on exposure of specific redox-active motifs to NO, or as a result of transnitrosation/transfer reactions from low-mass carrier SNOs or transfer from other protein SNOs. Under certain circumstances, SNOs could also serve as “NO donors” in the sense that they are activated by homolytic cleavage to form NO radicals which, in turn, diffuse to a site of bioactivity (2) .
Generally, specific protein thiols are targeted by S- nitrosylation. For example, of fifty (per receptor subunit) reduced cysteines in the ryanodine responsive calcium channel of skeletal muscle (RyR1), only one (Cys3635 ) is selectively S- nitrosylated to achieve calmodulin-dependent NO-mediated modulation of channel activity (44) . Excessive S- nitrosylation of RyR1 (at other cysteines) results in different bioactivity (44, 45) . Similarly, it is Cys69 , not other redox-active cysteines, that are S- nitrosylated on thioredoxin (24) .
Moreover, many SNO bioactivities are stereoselective; derivatives of the l -isomer of S- nitrosocysteine are highly active, whereas those of d -isomer of S- nitrosocysteine are inactive. Notwithstanding, both isomers are released as NO radicals (homolytically) at the same rate (17, 46) . Such stereoselective bioactivities include neural regulation––at the nucleus tractus solitarius––of ventilation, heart rate, and blood pressure (17, 46) , as well as regulation of peripheral and vascular smooth muscle tone (47) . These observations suggest the presence of specific S- nitrosothiol receptors; indeed, stereospecific L-CSNO antagonists have recently been developed.
Regulation of Gene and Protein Expression
The activities of a variety of nuclear regulatory proteins are affected dramatically by S- nitrosylation chemistry (Table 1⇓). These include hypoxia-inducible factor I (HIF-1) (41) , stimulating proteins 1 and 3 (Sp1 and Sp3) (48) , nuclear factor–κ B (NF-κ B) (49) , and the prokaryotic transcription factor OxyR (50–52) . The nuclear regulatory functions of S- nitrosylation appear to be dose-dependent, such that physiological levels of SNOs tend to sustain the transcription of physiological genes (48) , whereas supraphysiological or nitrosative-stress levels of SNOs induce the increased expression of stress response genes and proteins (49) . Higher levels of SNOs still may cause feedback inhibition of stress response protein transcription (50) . This dose-dependency is exemplified by both Sp1 and Sp3 levels. Concentrations of GSNO of 500 nM to 10 μ M increase Sp3 binding to the promoter and downstream transcription of the cystic fibrosis transmembrane regulatory gene, CFTR (48) . On the other hand, concentrations of GSNO in excess of 10 μ M inhibit Sp3 binding, augment competitive binding by Sp1, and prevent CFTR transcription (unpublished observations). Remarkably, physiological levels of GSNO (1–10μ M) are able dramatically to increase the expression and maturation of the most common human CFTR mutant, Δ F508 (42) . This increased expression and maturation allows for cell-surface expression of Δ F508 CFTR and allows for partial restoration of CFTR function in airway epithelial cells (53) . It appears that GSNO levels are low in the CF airway (54) . Thus, restoration of normal levels of GSNO in the CF airway epithelium appears to represent, at least in vitro, a “cure” for the defect.
S-nitrosylation of Transription Factors
HIF-1 is a heterodimer, composed of α and β subunits. The α subunit is constitutively transcribed and translated but is regulated by rapid degradation, whereas the β subunit is constitutively expressed. In response to hypoxia, HIF-1α is stabilized, which allows for its dimerization with HIF-1β and subsequent transcription of hypoxia-related genes coding for erythropoietin, heme oxygenase 1, and vascular endothelial growth factor (55) . It is believed that pO2 is sensed through prolyl hydroxylation to permit von Hippel Lindau protein (pVHL)-mediated ubiquitination of HIF-1α and its subsequent degradation by the proteasome (56, 57) . However, this mechanism of oxygen sensing has a threshold for responsiveness on the order of 1% oxygen (pO2 approximately 7 mm Hg). This level of pO2 has little relevance to the intravascular (renal or pulmonary) hypoxic signals, even in the most extreme cases of systemic hypoxemia. On the other hand, the deoxygenation of hemoglobin with ensuing transfer of SNO to AE1, resulting from an R → T conformational shift, occurs at physiologically relevant pO2 , owing to the dissociation of oxygen from oxyhemoglobin (14, 15) . Recent evidence suggests that, whereas NO radicals may inhibit the effect of hypoxia on HIF-1α stabilization, SNOs stabilize HIF-1α in normoxia and increase transcription of genes such as heme oxygenase 1 (42) . This normoxic activation of HIF-1 exhibits classical S- nitrosylation pharmacology in that it is: 1) not mimicked by 8-bromo cGMP, 2) not inhibited by oxyhemoglobin, 3) reversed by dithiothreitol, and, 4) as in the case of GSNO, is inhibited by the γ GT inhibitor, acivicin. Thus, the effects of nitrogen oxides on HIF-1 appear to be distinct. In normoxia, the DNA binding and activity of HIF-1 is increased through an S -nitrosylation event that impairs HIF-1α ubiquitination and degradation. This may occur by altering the interaction between pVHL and HIF-1α , such as through the S -nitrosylation of pVHL, or changing the proline hydroxylase activity that mediates the activation of HIF-1α . On the other hand, nitric oxide in hypoxia decreases HIF-1 binding and activity through a cGMP-dependent mechanism targeting the oxygen-dependent degradation and transactivating domains.
Both HIF-1 and NF-κ B increase the expression of NOS 2. NOS 2 activity feeds back to inhibit NF-κ B–mediated transcription by S- nitrosylating/inhibiting the p50 subunit of NF-κ B and by S- nitrosylating/inhibiting the inhibitor of NF-κ B (Iκ B) kinase (IKK) β , preventing phosphorylation of Iκ B and its dissociation-mediated activation of NF-κ B (49, 58) . However, these effects are not achieved until mid-to-high micromolar concentrations of SNOs are present.
OxyR is a thiol-containing transcription activator whose oxidation controls the expression of genes involved in hydrogen peroxide (H2 O2 ) detoxification. This prokaryotic transcription factor acts a redox–stress sensor recognizing both oxidation and nitrosation events (50) . In reduced form, OxyR has DNA binding activity but does not activate transcription (51) . However, in response to H2 O2 or S- nitrosothiols, OxyR induces the expression of a number of genes that protect E. coli from both oxidative and nitrosative stress (51) . OxyR monomers contain six cysteine residues. Two of these cysteine residues, Cys199 and Cys208 , are required for maximal activity. However, only C199 is essential for activity and it is flanked by an S- nitrosylation motif (H/R/K–C–D/E) (52) .
Regulation of protein function
A broad spectrum of membrane-associated proteins is S- nitrosylated. The activity of the NMDA receptor is decreased by S- nitrosylation of Cys399(18, 19) . Another example is anion exchange protein 1 (AE1) on the erythrocyte membrane, which is S- nitrosylated upon deoxygenation of membrane-associated hemoglobin (16) . However, the biology of cell signaling through SNO transfer reactions remains poorly understood. Although there is accumulating evidence that SNOs are stored in vesicles and released on cell stimulation (21, 35) , almost nothing is understood about the biochemistry, pharmacology, or physiology of these processes. Additionally, fundamental questions such as the identity of the putative stereoselective receptor for l -CSNO, and the mechanism by which SNO signals are transferred from AE1 across the endothelial cell membrane are not understood.
More is known, however, about the mechanisms by which S- nitrosylation may regulate cytosolic, mitochondrial, or extracellular proteins. Regulation of caspases-3 and -9 by release from the mitochondrial intermembrane space to the cytosol is described above. Detailed mechanisms have been described for S- nitrosylation-mediated functional regulation of several other proteins such as glyceraldehyde phosphate dehydrogenase (36, 37, 59) . Cytosolic S- nitrosylation reactions also appear to affect the regulation of transition metal homeostasis. For example, S- nitrosylation of metallothionein results in release of free cytosolic Zn2+(60) . However, as noted previously, the cytosol is a highly reducing environment, and cytosolic reservoirs such as SNO-thioredoxin and GSNO appear to be tightly regulated. Extracellular (circulating and interstitial) proteins are also S- nitrosylated (3, 7) . Some of these proteins may serve as nonspecific transport vectors and reservoirs; others may be functionally modified.
Perhaps the most intriguing and controversial protein modification is S- nitrosylation of hemoglobin. There is uniform agreement with Stamler’s original hypothesis that hemoglobin is endogenously S- nitrosylated at the β 93 cysteine (14) . Circulating levels of SNO-hemoglobin are on the order of 200–500 nM. One measurement technique involving sample pretreatment with high concentrations of cyanide, in addition to other, indirect evidence, suggests that iron-to-thiol transfer does not occur during T → R transformation and that SNO exposure on the hemoglobin surface in T confirmation hemoglobin does not favor transfer of the NO moiety to AE1 or other thiols. On the other hand, these same allosterically regulated transfer reactions have been demonstrated to occur by 1) four separate biochemical techniques (i.e., mass spectrometry, photolysis, chemiluminescence, reductive chemiluminescence and reduction fluorescence) (14, 15, 17) ; 2) three separate bioassays (14, 17, 61) ; and 3) computational modeling. Additional mechanistic validation has recently been provided using electron spin resonance spectroscopy (62) . The paradigm suggested by this body of evidence from several different research groups is that hemoglobin deoxygenation allows transfer of NO from SNO–hemoglobin to relax medium-sized resistance vessels (augmenting blood flow to hypoxic tissues), stimulating neural sensation of hypoxia to increase ventilation, and increasing hypoxia-associated transcription in physiologically relevant hypoxia. This paradigm is physiologically and teleologically appealing and is supported by recent work on human hypoxia signaling in vivo (63) ; however, hemoglobin chemistry is complex, and much work remains to be done. A measurement technique involving sample pretreatment with high concentrations of cyanide, strong acids and, triiodide has been used to argue against FeNO–SNO transfer in hemoglobin (20) . However, this method cannot differentiate between FeNO–SNO and cannot accurately measure either in a complex biological system. Furthermore, methodology notwithstanding, the measurement cannot be used to argue for or against allostery, as allostery cannot be probed by such a measurement. Thus, one would not anticipate an SNO gradient.
SNO signaling is not exclusively relevant to mammalian physiology. A variety of intriguing observations have been made regarding the role of S- nitrosylation chemistry in viral replication (inhibition of cysteine proteases can prevent replication), hemoglobin-mediated oxygen scavenging in Ascaris, and bacterial stress responses (50, 64–66) . These subjects are being actively studied in anticipation that they will lead to new therapeutic strategies for a variety of infectious diseases.
Therapeutic Implications
Inhaled NO may exert some of its salutary activities through cGMP-independent reactions in the airway epithelium, pulmonary vascular smooth muscle, and airway smooth muscle (3, 66–70) . Recent observations regarding S- nitrosoglutathione–mediated cell signaling reactions have been exploited to develop new therapeutic agents. One such agent is ethyl nitrite, an S- nitrosylating agent that does not generate NO, which is a more potent and effective treatment for pulmonary hypertension than NO (6, 71) . Similarly, whereas inhaled NO is not effective as a treatment for cystic fibrosis (72) , inhaled S- nitrosoglutathione appears to be effective in vitro and in vivo (42, 72) . Several new therapies making use of this chemistry are being investigated.
Summary
Endogenous S- nitrosylation reactions signal a broad spectrum of cellular activities independently of NO radical formation/guanylyl cyclase activation. These include transcriptional and post-transcriptional regulation of protein expression as well as regulation of membrane, cytosolic, mitochondrial, nuclear, and extracellular protein functions. The cellular synthesis, compartmentalization, and catabolism of low-molecular weight and protein S- nitrosothiols appear to be specifically regulated; however, the study of each of these topics is in its infancy.
Acknowledgments
This work was supported by NIH/NHLBI: HL59337, HL69170, 1U19-A134607 (BG); and NIH/NHLBI: HL68173-01 (LP), NIH/NICHD K12HD01421-01 (AD and JC).
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Lisa Palmer, PhD , (left) is an Associate Professor of Research in the Departments of Pediatrics and Anesthesiology at the University of Virginia. Her research interests include nitrogen oxide– and hypoxia-induced regulation of the transcription factor HIF-1 and pulmonary hypertension. Ben Gaston, MD , (second from left) is an Associate Professor of Pediatrics in the Division of Respiratory Medicine in the Department of Pediatrics at the University of Virginia. His research interests include studies on nitrogen oxide metabolism and lung inflammation. Jeannean Carver, MD , (second from right) is an Assistant Professor of Pediatrics in the Division of Pediatric Critical Care in the Department of Pediatrics at the University of Virginia. Her research interests include intracellular immune-cell signaling and septic shock. Allan Doctor, MD , (right) is an Assistant Professor of Pediatrics in the Division of Pediatric Critical Care at the University of Virginia. His research interests include nitrosative signaling in the pulmonary microcirculation in the setting of lung injury and respiratory failure. Please address correspondence to LAP. E-mail lap5w{at}virginia.edu; fax (434)-982-4927.

