Estrogen Signaling: A Subtle Balance Between ERα and ERβ
Abstract
The biological actions of estrogens are mediated by estrogen binding to one of two specific estrogen receptors (ERs) ERα and ERβ, which belong to the nuclear receptor superfamily, a family of ligand-regulated transcription factors. ERα and ERβ are products of different genes and exhibit tissue- and cell-type specific expression. The characterization of mice lacking ERα, or ERβ, or both has revealed that both receptor subtypes have overlapping but also unique roles in estrogen-dependent action in vivo. Additionally, ERα and ERβ have different transcriptional activities in certain ligand, cell-type, and promoter contexts. Both receptors, however, are coexpressed in a number of tissues and form functional heterodimers. The biological roles of ERα /β heterodimers in the presence of each respective homodimer are unknown. When coexpressed, ERβ exhibits an inhibitory action on ERα -mediated gene expression and in many instances opposes the actions of ERα. A number of ERα and ERβ isoforms have also been described, many of which alter estrogen-mediated gene expression. Uncovering the molecular mechanisms regulating the expression of both ERs, and how ERα and ERβ directly or indirectly affect each other’s function are paramount to understanding the cellular and biological events of estrogen-mediated gene regulation in normal and diseased tissues.

Introduction
Estrogens regulate many physiological processes, including normal cell growth, development, and tissue-specific gene regulation in the reproductive tract and in the central nervous and skeletal systems (1, 2) . Estrogens also influence the pathological processes of hormone-dependent diseases, such as breast, endometrial, and ovarian cancers, as well as osteoporosis (2) . The biological actions of estrogens are mediated by binding to one of two specific estrogen receptors (ERs), ERα or ERβ , which belong to the nuclear receptor (NR) superfamily, a family of ligand-regulated transcription factors (1) . Ligand-binding induces conformation changes in the receptor leading to dimerization, protein–DNA interaction, recruitment of coregulator proteins, and other transcription factors, and ultimately the formation of the preinitiation complex (3) . ERs regulate gene expression by binding to their cognate response element or through protein–protein interactions with other transcription factors (4) . The discovery in rat prostate of ERβ (5) and its subsequent cloning in other species (6) has caused a paradigm shift in our understanding of estrogen action.
ERα and ERβ contain the evolutionarily conserved structural and functional domains typical of NR family members, including domains involved in DNA-binding, dimerization, ligand binding, and transcriptional activation (7) (Figure 1⇓). ERα and ERβ share a high degree of sequence identity within their DNA-binding domains (DBDs). The amino acid sequence of the P-box, a motif within the DBD critical for receptor–DNA recognition and specificity, is identical between the two receptors. Thus, it is not surprising that both receptors bind estrogen responsive elements (EREs) with similar specificity and affinity, although differential subtype affinities and responses to a subset of natural EREs have been described (8) . The ligand-binding domains (LBDs) are also conserved and both receptors exhibit similar affinities for the endogenous estrogen, 17β -estradiol (E2). However, ERα and ERβ exhibit different affinities for some natural compounds and novel subtype-specific ligands have been reported (9, 10) . Characterization of mice lacking either ERα , or ERβ , or both (α ERKO, β ERKO, and αβ ERKO, respectively) has demonstrated that each subtype has similar but also unique roles in estrogen action in vivo. Both ERs are widely distributed throughout the body, displaying distinct but overlapping expression patterns in a variety of tissues (1, 2) . ERα is expressed primarily in the uterus, liver, kidney, and heart, whereas ERβ is expressed primarily in the ovary, prostate, lung, gastrointestinal tract, bladder, and hematopoietic and central nervous systems. ERα and β are, however, coexpressed in a number of tissues including the mammary gland, epididymis, thyroid, adrenal, bone, and certain regions of the brain. Although both ER subtypes may be expressed in the same tissue, they might not be expressed in the same cell type. Nonetheless, ERα and ERβ proteins have been simultaneously detected in many cell types including neurons and thymocytes (11, 12) , and these as well as other cell types that coexpress both ER subtypes are targets for heterodimerization and potential interplay between the two receptors.
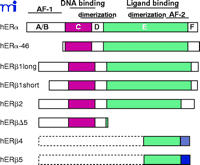
Schematic representation of human estrogen receptor α and β and their respective isoforms. Note that both hERβ2 and hERβΔ5 can have either the hERβ1 long or the short N-termini. In addition, there is no evidence of full-length hERβ4 or hERβ5. The different fill patterns of the 3′ end of hERβ2, 4, and 5 represent the divergent C-terminal regions of these isoforms.
The transactivating functions of ERα and ERβ are mediated by two separate but not mutually exclusive transcription activation functions (AFs) that allow the receptors to stimulate the transcription of estrogen-regulated genes: an N-terminal ligand-independent activation function (AF-1), and a C-terminal ligand-dependent activation function (AF-2) located within the LBD (7) . The AFs contribute to estrogen-mediated transcription and mediate cell- and promoter-specificity. A comparison of the AF-1 domains of the two ERs has revealed that this domain is very active in ERα on a variety of estrogen responsive promoters, but under identical conditions, the activity of AF-1 in ERβ is minimal (13) . Furthermore, these two receptors exhibit distinctive responses to the synthetic antiestrogens tamoxifen and raloxifene. For example, these ligands are partial ER agonists for ERα but act as pure ER antagonists for ERβ (13) . The differences between the respective N-terminal regions of the ERs have been suggested as a possible explanation for their diversity of responsiveness to several ligands. The AF-2 mediated transcriptional activities of ERs are dependent on interactions with and recruitment of cofactor proteins to estrogen responsive promoters. Cofactors can be classified as coactivators, which promote NR activity, or corepressors, which attenuate NR activity.
Crystallographic studies have revealed that the LBDs of both ERα and ERβ share a similar overall architecture and have demonstrated that the AF-2 interaction surfaces are composed of amino acids in helices 3, 4, 5, and 12, and that binding of ligands alters the position of helix 12. When the ERα LBD is complexed with agonists such as E2, helix 12 is positioned over the ligand-binding pocket and forms an interaction surface for the recruitment of coactivators. In contrast, when either the ERα - or ERβ -LBDs are complexed with antagonists, helix 12 is displaced from its agonist position and occupies the hydrophobic groove formed by helices 3, 4, and 5, causing helix 12 to disrupt the coactivator interaction surface (14, 15) . Functional and structural studies have shown that coactivators interact with the AF-2 region via short leucine-rich motifs (i.e., LxxLL) termed “NR boxes” to transduce ligand signals to the basal transcriptional machinery. These motifs represent the primary docking sites to the AF-2 domain and exist in many cofactors (16) . A large subset of these proteins include the steroid receptor coactivator (SRC) family (SRC1, SRC2/TIF2/GRIP1, and SRC3/AIB1/RAC3) (17) and components of the mammalian mediator complex [thyroid receptor associated proteins (TRAP), vitamin-D receptor interacting proteins (DRIP), activator-recruited cofactor (ARC)] (3, 18) , which possess chromatin-remodeling ability and tether activated receptors to the basal transcriptional machinery (3) . Additional cofactors, such as the histone acetyltransferase (HAT), CREB-binding protein (CBP), the highly related p300, and the histone methyltrasferases CARM-1 and PRMT1 are tethered to the ERs through interactions with the SRC family of coactivators (3, 18) . The recruitment of cofactors with histone modifying and chromatin remodeling activities by ERs overcomes the repressive features of chromatin leading to active transcription through the general transcriptional machinery (19) .
In the absence of ligand, some NRs associate with the NR corepressors, namely, silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) and nuclear receptor corepressor (NCoR) (18) . Both SMRT and NCoR recruit SIN3 and histone deacetylases (HDACs) to form a large corepressor complex that contains histone deacetylase activity, implicating histone deacetylation in transcriptional repression (20).
It has been suggested that corepressors and deacetylases are recruited to ER target genes where they antagonize coactivator and HAT complexes. Indeed the histone deacetylase activity recruited by corepressor complexes such as NCoR–SIN3–HDAC2 is required for the transcriptional repression of tamoxifen-bound ERα , and the loss of corepressors might be one mechanism of tamoxifen resistance (20).
Although ERα and ERβ share similar mechanisms of action, several differences in the transcriptional abilities of each receptor as well as distinct phenotypes between gene-null animals have been identified, suggesting that these receptors may regulate distinct cellular pathways. When ERs are coexpressed, ERβ exhibits an inhibitory action on ERα -mediated gene expression (4, 21, 22) . A number of isoforms of ERα and ERβ have also been described, many of which also alter estrogen-mediated gene expression (23, 24). Uncovering the molecular mechanisms regulating the expression of both ERs, and the complex interplay between the two receptors is paramount in understanding the cellular and biological events of estrogen-mediated gene regulation. This review focuses on the molecular mechanisms that regulate expression of the ERα and ERβ genes and how ERα and ERβ directly or indirectly affect the expression of the other subtype.
ERα and ERβ Isoforms
ERα and ERβ are products of distinct genes on different chromosomes. ERα is located at chromosomal locus 6q25.1 (25) , whereas ERβ is found at position 14q22–24 (26) . Several ERα and ERβ splicing variants have been described, but whether all transcripts are expressed as functional proteins and have biological functions remains unclear.
The generation of human ERα mRNA transcripts is a complex process that involves at least seven different promoters and exhibits cell line-dependent promoter usage [reviewed in (27) ]. Most ERα variants differ only in the 5′ UTR and result in the expression of the full-length 66-kDa form of ERα (27) . Flouriot and colleagues have, however, identified a shorter 46-kDa isoform of ERα generated from an internal ATG start codon (28) (Figure 1⇑). The shorter 46-kDa isoform of hERα lacks exon 1 and consequently the N-terminal AF-1 region (28) . This isoform is present in human osteoblasts and in the breast cancer cell line MCF-7, and heterodimerizes with wild-type ERα , thereby suppressing its AF-1–dependent transcriptional activity. This inhibition was observed in HepG2 cells, where ERα activity is primarily mediated through AF-1, but not in HeLa cells in which estrogen responses are predominantly mediated through AF-2 (28) . Although AF-1 specific repression mediated by the 46-kDa ERα isoform might be necessary for cell proliferation, much of the identification and characterization has been performed in cell-lines and this isoform has not yet been identified in human tissue samples. Therefore, its precise role in modulating the pleiotrophic effects of estrogen remains to be determined.
The first characterization of the ERβ promoter identified a region referred to as exon 0N (29) , and subsequent analysis identified a second upstream promoter region referred to as exon 0K (30) . The Human Genome Project has aided investigators in identifying at least five additional exons located between the 0K and 0N regions, suggesting that ERβ is under sophisticated regulatory control similar to that reported for ERα (31) . In addition, several alternative splicing variants of ERβ , some with extended N-termini and others with truncations and/or insertions in the C-terminal LBD have been reported (26) (Figure 1⇑). The expression levels of many of these isoforms are higher in human breast tissues than that of wild-type ERβ , and there are data supporting the protein expression of several ERβ isoforms (32, 33) . The original human ERβ clone encoded a protein of 485 amino acids (6) ; however, cloning of an additional N-terminal sequence has extended the N-terminus resulting in a 530 amino acid protein (34) . These proteins have been designated ERβ 1 long (530 aa) and ERβ 1 short (485 aa), and the ERβ 1 long form is currently regarded as the full-length wild-type ERβ . Although some transfection studies suggest that both forms are functionally equivalent (35) , the possibly distinct roles of the variable N-termini have yet to be fully explored. Wilkinson and colleagues have described the identification of an putative upstream in-frame initiation codon in hERβ , yielding a predicted protein of 548 amino acids; unlike ERβ 1, the ERβ 548 amino-acid isoform exhibits tamoxifen and raloxifene partial-agonist responses on an ERE driven reporter gene (36) . However, a recent study failed to detect hERβ 548 in samples from African, Caucasian, and Asian populations (37) , therefore questioning the physiological relevance and importance of this isoform.
ERβ variants lacking exons 5 and/or 6 have been identified in human cancers and various cell lines (38) . Loss of exon 5 results in a codon frameshift and produces a truncated receptor that lacks the majority of the LBD. Coexpression of this isoform with either wild-type ERβ 1 or ERα inhibits E2-dependent ERE-driven gene activity; thus, this isoform may function as an ER inhibitor in vivo. Another ERβ isoform, ERβ 2 (also called ERβ cx) (24) , is identical to the ERβ 1 long form except that 26 unique amino-acid residues, termed exon 9, replace the amino acids of exon 8. ERβ 2 does not bind ligand, nor does it activate the transcription of an estrogen-sensitive reporter gene. Furthermore, ERβ 2 shows preferential heterodimerization with ERα rather than with ERβ , inhibiting ERα DNA binding and having a dominant-negative effect on ligand-dependent ERα reporter gene activity (24) . Moreover, hERβ 2 inhibits the transcriptional activity mediated by raloxifene-ERα complexes on the raloxifene responsive tumor growth factor–beta 3 (TGF-β 3) promoter, whereas no transcriptional inhibition was observed with ERβ 1 on raloxifene–ERα complexes (39) . These data suggest that the ERβ 2 and ERβ 1 isoforms may differentially modulate estrogen action. Several additional ERβ isoforms have been reported, although full-length sequences have not been determined (40) (Figure 1⇑). Some isoforms exhibit species-specific expression; for example, an ERβ variant in rat termed ERβ 503 containing an eighteen amino-acid insertion in the LBD has not been detected in humans. ERβ 503 and ERβ 1 heterodimerize and bind to consensus EREs but the affinity of many ligands for ERβ 503 is 1000-fold lower than for ERβ 1 (41).
Regulation of ERα and ERβ Expression Levels
The relative expression levels of ERα and ERβ are significantly altered during the development of breast cancer (42,43) ; however, the molecular details surrounding the receptors’ regulation remain poorly understood.
The mechanisms that regulate ERα mRNA levels have been best studied in the context of primary breast carcinoma and breast cancer cell lines, because ERα participates in the initiation and progression of this type of neoplasia. Transcriptional enhancer elements located in the 5′ UTR of the ERα promoter have been identified and shown to contribute to the differential expression of ERα in breast cancers. Two coregulator proteins, activator protein 2-γ (AP2-γ ) and estrogen receptor promoter B associated factor (ERBF-1), are partly responsible for the enhancement of ERα expression (44, 45) ; however, AP2-γ is not expressed in all breast tumor cells and ERBF-1 specifically enhances the expression of ERα from a distal promoter region that may not be utilized in all cell types (44) .
The expression level of ERα is also intricately regulated through DNA methylation and chromatin condensation of promoter regions via hypermethylation of CpG islands in the ERα promoter. Hypermethylation of the ERα promoter is associated with a marked decrease in ERα mRNA expression among ERα -expressing cancer cell lines, and the inhibition of DNA-methyltransferases reactivates ERα expression in these cell lines (46) . Increased CpG methylation of the ERα promoters is reported to be significantly more extensive in BRCA1 -linked human tumors, suggesting that inhibition of this process may be of particular benefit to patients that inherit a mutation in BRCA1 (47) .
The molecular mechanisms that govern the regulation of ERβ expression are relatively unclear (29) . To date, mRNA transcripts that include the 0K versus the 0N exon are expressed in differing amounts in different cell lines (and types) though whether the five (thus far described) putative promoter regions exhibit a similar cell-type-specific usage and whether these transcripts encode receptors that differ from wild-type receptor remains to be determined. Several response elements have been identified in the 5′ UTR of the ERβ promoter, but no specific transcription factor has been identified that might contribute to the differential expression of ERβ protein. As in the case of ERα , the ERβ promoter regions are also subject to DNA methylation. Of particular note is the ERβ 0N promoter, which is methylated in a number of prostate cancer cell lines and carcinogenic tissue samples (48) . This may have important clinical implications because ERβ is the dominantly expressed ER in the prostatic epithelium and β ERKO mice display age-related prostatic hyperplasia (49) . Ongoing studies from our group indicate that the 0K and 0N promoters are differentially methylated in a wide range of human breast cancer cells, supporting the findings that DNA methylation is an important regulatory mechanism of ERβ expression levels.
The Expression ERα and ERβ is Autoregulated
Several groups have demonstrated that exposure to estrogen can modulate the expression of ERα and ERβ (50–52) , but little is known about the biological importance and the molecular mechanisms involved in this autoregulation. In the case of ERα , exposure to E2 results in a ligand-dependent increase in the activities of two distinct promoter regions, although it is not known whether all of the distinct ERα promoters are responsive to estrogen (51, 52) . It has been hypothesized that autoregulation may contribute to ERα overexpression in some breast tumors (52) . Similarly, little is known about the potential ERα -dependent regulation or autoregulation of the ERβ promoters. Our group and others have observed a time-dependent estrogen-induced increase in ERβ mRNA in the human breast cancer cell-line T47D [(38) (unpublished results)]. Subsequent chromatin immunoprecipitation (ChIP) assays have revealed the time-dependent recruitment of ERα to an ERE half-site in the ERβ 0N promoter (unpublished results). Interestingly, an ERE half-site has also been identified in the 0K exon though it is unclear whether ERα or ERβ regulates this promoter. In contrast, analyses of neuronal tissue from α ERKO and from wild-type mice show that estrogen treatment tends to decrease the number of ERβ -immunoreactive cells in wild-type mice, whereas such cells were increased α ERKO mice (50) . Moreover, long-term exposure of primary endothelial cells to E2 increases the expression of ERα but decreases that of ERβ (53) . Taken together, these studies highlight the complex interplay between ERα and ERβ in the regulation and autoregulation of their respective promoters and suggest that this regulation exhibits cell-type and tissue variation.
ERβ Opposes ERα-Mediated Transcription
ERβ appears to act as a dominant regulator of estrogen signaling, and when coexpressed with ERα , ERβ causes a concentration-dependent reduction in ERα -mediated transcriptional activation (21, 54) . These antagonistic effects of ERβ on ERα might arise from differences in their respective transactivation regions (54) . ERα activation requires the combination of the two AFs for synergistic transcriptional activation, but the individual regions exhibit independent activity in a cell-type– and promoter-dependent manner. Both subtypes contain the potent C-terminal AF-2 (7) , but unlike ERα , ERβ contains a weaker N-terminal AF-1, which might possess repressive activity (35) .
ERs can regulate gene expression through several different modes including direct DNA binding (as homodimers or as heterodimers) or through tethering to other transcription factors such as activating protein-1 (AP-1) and stimulating protein 1 (Sp1) (Figure 2⇓). In many instances ERα and ERβ exhibit opposing actions in the regulation of several promoters and specific response elements. Tethered to AP-1, ERα exhibits E2-dependent activation of transcription at AP-1 sites, whereas E2-bound ERβ has no effect (4) . Raloxifene binding to ERβ causes an increase in reporter gene expression, whereas binding to ERα results in minimal activation examined under identical conditions (4) . The ERα –AP-1 complex mediates the estrogen-dependent activation of the progesterone receptor promoter (55) .
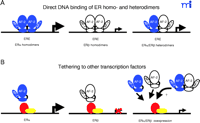
Alternative estrogen-dependent transcriptional regulation by ERα –ERβ homo- and heterodimers. The different modes of direct DNA binding of ER subtypes either as homo- or heterodimers to estrogen response elements (EREs) (A). Representation of the transcriptional regulation by ER subtypes through tethering to other transcription factors, such as activating protein 1 (AP-1) and stimulating protein 1 (Sp1) (B). The relative estrogen-dependent transcriptional activities are represented by the magnitude of the arrow. ERα and ERβ are represented in blue and white, respectively.
ERα and ERβ oppose each other’s function in the regulation of the cyclin D1 promoter (21) . Unlike ERα , E2-bound ERβ represses cyclin D1 expression and blocks ERα -E2–mediated induction when both receptors are present. The increased expression of the cyclin D1 gene participates in estrogen-provoked proliferation in vivo, and the cyclin D1 protein is overexpressed in over 50% of mammary carcinomas. Several groups have shown that ERα and ERβ form functional heterodimers in vitro and in vivo and that if both isoforms are expressed, the heterodimers predominate (56) . The role that heterodimers play in estrogen signaling, specifically in mediating the antagonistic effects of ERβ on ERα transcriptional activity, is unknown.
ERs also modulate target-gene expression through interaction with the transcription factor Sp1 (57) . Both ERα and ERβ specifically interact with Sp1, and agonists as well as antagonists activate the ERα –Sp1 complex; however, little, if any, increase in transcriptional activation is observed for Sp1-associated ERβ complexes (57) . This differential responsiveness for each subtype has been mapped to the AF-1 domains––in domain swapping experiments, complete reversal of phenotypes was observed (57) . Furthermore, coexpression of ERα with increasing concentrations of ERβ results in repression of ERα –Sp1 activity, supporting a functional antagonism between ERβ and ERα .
It may be argued that the ERβ -dependent antagonism of ERα -mediated responses is restricted to a limited number of genes and does not represent a general mechanism in ER signaling. Nonetheless, global antagonism by ERβ on ERα transcriptional activity is supported by a number of recent studies describing ERβ -directed repression of several ERα -mediated effects including fat reduction and cellular proliferation in the uterus and prostate (58) . In addition, we have applied high density microarray analysis of bone and liver tissue isolated from α ERKO, β ERKO, or αβ ERKO animals, and the data support the repressive effects of ERβ on ERα -mediated transcriptional activity (22) . In the absence of ERα , ERβ partially mediates the effect of estrogen on gene transcription. Collectively, data from cell-based assays, gene expression studies, and analysis of ER null animals strongly support the contention that ERβ attenuates ERα activity.
Recruitment of Subtype-Selective Cofactors
There has been a strong focus on the cloning of a large number of NR coactivators and corepressors and the characterization their respective enzymatic activities (18) . These proteins participate in the determination of target-gene expression and act to bridge activated receptors to the basal transcriptional machinery. The precise roles of each type of coactivator, as well as the functional interactions among them, during NR-dependent transcription have not been well defined. Altered expression of ER cofactors has been reported during human breast tumorigenesis (59) , and selective cofactor recruitment by ERα and ERβ may serve as a mechanism for differential transcriptional activities between the two receptors.
A number of proteins have been isolated that interact in a ligand-dependent manner via their NR-box sequences with the AF-2 region of ERs. The thyroid receptor–associated protein 220 (TRAP220), a component of the mediator complex, which bridges ERs to RNA polymerase II, interacts with both ERα and ERβ but exhibits selective affinity for ERβ (60) . This subtype selective affinity is influenced by the respective C-termini, the F domains (61) . The SRC family members are important mediators of AF-2–dependent responses and have been shown to interact in vivo with both ERs (17) . SRC-3/AIB1/RAC3 is overexpressed in a variety of hormonally regulated cancers including breast, uterine, and prostate cancers (62) , and overexpression of cofactors is suspected to contribute to the enhancement of estrogen-dependent tumorigenesis (59) . Both SRC-2/TIF2/GRIP1 and SRC-3/AIB1/RAC3 enhance ERα -mediated transcription in a cell-type specific manner (63, 64) . The affinities of ERα and ERβ for different coactivators and various NR boxes present within coactivators also display ligand-dependent variability (65) . For example, in the presence of E2, SRC-1 or SRC-2/TIF2/GRIP1 can be recruited to both ER subtypes with similar affinity, whereas in the presence of genistein, both SRC coactivators are more strongly recruited to ERβ than ERα (65) .
The contexts of response elements and cell-type specificity affect the actions of regulatory proteins, such as coactivators. Although the mechanisms are not understood, it is clear that within a regulatory protein complex, a given factor may activate transcription, repress it, or have no regulatory activity. Different response elements induce alterations in the efficiency of cofactor–AF-2 interactions thereby inducing differential binding of SRC coactivators to ERα or ERβ in the presence of diverse ERE elements (66) . These differences may serve as a basis for promoter-specific activities between the two ER subtypes. In addition, the activity of SRC-2/TIF2/GRIP1 can alter between that of a coactivator and a corepressor depending on the response element (67) . On the collagenase-3 gene, SRC-2/TIF2/GRIP1, but not SRC-1 or SRC-3/AIB1/RAC3, enhances glucocorticoid receptor–mediated transcriptional repression (67) . These data suggest that differential recruitment and/or variability in the expression levels of coactivators can play a significant role in mediating transcriptional differences between ERα and ERβ .
Subtype-Specific Target Genes
ERα and ERβ exhibit similar affinity for different EREs (8) , which is not surprising given the high degree of sequence identity between their DBDs. Thus, to date, subtype-selective EREs have not been described, although there are examples of subtype-selective interactions at other response elements, such as SFREs [steroidogenic factor 1 (SF-1) response elements]. Laudet and colleagues have explored the potential interplay among ERα , ERβ , and members of the estrogen receptor related receptors (ERRs) (68) . They have shown that in addition to binding EREs, ERα and ERRs also bind to SFRE whereas ERβ does not. Despite the ability of ERα and ERβ to form heterodimers, ERα is unable to drive ERβ to the SFRE of an artificial reporter or to the SFRE-containing osteopontin promoter (68) . This suggests that ER subtype-specific regulation might preferentially take place with response elements other than those with an ERE background, or perhaps, through tethering to additional transcription factors, as is observed at AP-1– or Sp1-responsive elements (4, 57) . However, different EREs cause distinct conformational changes in receptors that are independent of ligand-induced effects, and these changes in turn influence interactions with cofactors (66) . Because the ERE sequence directly influences the amount of ER bound to DNA and indirectly influences the affinity of ER for coactivators, the context of the ERE may also contribute to subtype-specific gene regulation. Moreover, in light of the recent findings on the influence of chromatin on ERβ activity [see below, (69) ], it is conceivable that although both subtypes are able to interact with an ERE in vitro, this interaction may not result in the formation of an active complex on chromatin templates in vivo.
The identification of subtype-specific target genes will be important for understanding the biological functions of each subtype but it will also present investigators with a significant challenge. We and others have approached this problem by applying high-density microarray analysis to tissues isolated from ER-null mice (as well as to cell lines that express both receptors) followed by extensive bioinformatics to identify not only new regulatory pathways and new target genes, but also possible subtype-specific target genes. This strategy is central in clarifying the complexities of estrogen receptor signaling networks.
Influence of Chromatin on ER Transcriptional Activity
In eukaryotes, DNA is associated with histones and organized into nucleosomes, which together form chromatin. The packaging of genomic DNA into chromatin restricts access of the transcriptional machinery, thereby causing a repression in transcription. Although chromatin was once overlooked and considered a passive player in transcriptional regulation, the significant role that chromatin has in the proper activation and repression of transcription has been demonstrated experimentally in a number of in vitro and cell-based assays (70) . The basic N-termini of histones are subject to a number of posttranslational modifications, including lysine acetylation, lysine and arginine methylation, serine phosphorylation, and ubiquitinylation (71) . These modifications form the basis for the “histone code hypothesis,” which predicts that modifications of a histone tail influence the regulatory features of target loci (72) .
Recently, the field of transcriptional regulation has witnessed a rapid emergence of the ChIP assay, which is becoming a routine technique for studying the temporal regulation of chromatin templates and has helped to uncover new mechanisms of transcriptional regulation. ChIP assays offer the powerful advantage of studying transcription factor binding to endogenous promoters in vivo under different physiological conditions. Two independent laboratories have investigated the dynamics of ligand-dependent cofactor recruitment by ERα to estrogen responsive promoters (73, 74) . Their data demonstrate that ERα reaches maximal occupancy of the cathepsin D and pS2 promoters after forty-five to sixty minutes of treatment, although significant promoter occupancy is also apparent minutes after exposure to E2 [(74) and our unpublished data]. This may not be surprising because significant levels of ERα are bound to promoters in the absence of ligand (73, 74) . Following the initial peak of binding occupancy, ERα leaves the promoters and returns in a second peak after 120 to 150 minutes of treatment (73, 74) , suggesting that ERα cycles on and off the promoter during active rounds of transcription. It has been demonstrated that recruitment of E3 ligases, enzymes that covalently add one or more ubiquitins to target proteins, and subunits of the 19S proteasome are necessary in the cycling of ERα to promoters (75) . ChIP data also support the sequential formation of complexes of different coregulators during the onset of transcription; however, there are inconsistencies concerning the simultaneous recruitment of specific cofactors, namely the mediator and p300/CBP–SRC complexes (73, 74) . Additionally, the kinetic profiles were not sufficient to address the extent of cycling that occurs during promoter activation or the mechanisms and participants involved in repression of these “activated promoters.” Moreover, little is known about temporal recruitment of ERβ to estrogen responsive promoters and also about the potential role ERβ may exert on dynamics of ERα -mediated gene expression. ERβ has been conclusively demonstrated to have an antagonistic effect on ERα activity, and ChIP assays offer a powerful means of investigating the effects of ERβ on ERα and cofactor recruitment during the activation and shut down of transcription on endogenous promoters.
Kraus and colleagues have recently investigated the potential impact of chromatin on ERα and ERβ -mediated transcriptional activities using a well-established in vitro chromatin assembly assay (76) . They have elegantly shown that complexes of the mediator, CBP/p300, and SRC are required for maximal transcriptional enhancement of ERα -dependent chromatin templates and that the mediator has distinct functions during transcriptional activation as compared to the p300/CBP–SRC complex (77) . The p300/CBP-SRC complex was primarily involved in the stable formation of the preinitiation complex in the initial round of transcription, whereas the mediator was not only involved in the first round of complex formation but also in subsequent rounds of reinitiation (77) . Perhaps more intriguing was the finding that ERα and ERβ exhibited similar abilities to activate transcription on naked DNA, but that ERα is much more efficient at activating transcription on chromatin templates (69) (Figure 3⇓). The difference is attributable to the N-terminal AF-1 region of ERα because the inability of ERβ to function on chromatin templates was overcome by swapping its AF-1 region with that of ERα . The studies of Kraus and colleagues suggest that the AF-1 regions of the ERs harbor distinct features affecting the recruitment of cofactors, which function on genomic templates and not naked DNA. In agreement with other reports, coexpression of ERα and ERβ results in repression of ERα -mediated transcription (69) .
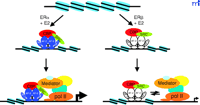
The influence of chromatin on the transcriptional activities of ERα and ERβ. Both ER subtypes exhibit comparable activity on naked DNA, but differential activity on chromatin templates. ERα strongly activates transcription on chromatin templates, whereas chromatin structure is very restrictive for transcription mediated by ERβ. These differences may be due to the inability of ERβ to recruit a stable preinitiation complex to chromatin templates, demonstrating that chromatin represents an important regulatory aspect of ER-mediated gene expression.
Relatively few proteins appear to bind primarily to the N-terminal AF-1 region of ERs. For example, the RNA-binding protein p68/72, which is found in a complex containing SRC coactivators and the RNA coactivator SRA, binds selectively to ERα (78) . Interestingly, both p68/p72 and TATA-binding protein (TBP) show selectivity for the N-terminal A/B region of ERα (78, 79) . Screening for novel subtype specific AF-1 interaction partners, and further biochemical and cell-based assays will be necessary to uncover the molecular mechanisms behind the differential activities of ERα and ERβ on chromatin templates.
Plasma Membrane Estrogen Receptors
Increasing evidence suggests that distinct pools of ERs that localize to the plasma membrane play important roles in estrogen-dependent responses. Several groups have demonstrated the E2-induced rapid generation of a number of signaling cascades including cyclic AMP, inositol-1,4,5-trisphosphate (IP3 ), phospholipase C, and members of the MAP kinase family [reviewed in (80) ]. Many of these effects are thought to arise through the activation of signaling cascades by distinct pools of ERs, which locate to the caveolae of the plasma membrane. Transient transfection of both ER subtypes from single cDNAs results in a small but significant pool of receptors localizing to the plasma membrane, suggesting that the plasma membrane pool of ERs arises from the same gene product as that localized to the nuclear compartment (81) . The precise mechanism and structural features of ERs that result in membrane localization have not been determined. Recently, however, Ser522 of ERα has been shown to be necessary for the efficient translocation and function of ERα at the plasma membrane; mutation of this residue to alanine significantly reduces ERα translocation to the plasma membrane (82) . Interestingly, Levin and colleagues have also reported the differential regulation of c-Jun kinase by the different membrane pools of E2 bound ERα and ERβ ; whereas ERβ activated c-Jun N-terminal kinase (JNK), ERα inhibited its activation (81) . The E2-dependent inhibition of JNK activation has been suggested as a mechanism for breast cancer survival since the inhibition of JNK prevents the inactivating phosphorylation of Bcl2 and the subsequent activation of the caspases resulting in cell death (83) . These data demonstrate that opposing actions of ER subtypes not only occur at the transcriptional level but also in the plasma membrane pool of ERs, which could have significant consequences for estrogen-dependent cell biology.
Conclusions and Future Prospects
Despite being discovered almost ten years after ERα , ERβ appears to be the dominant player in a number of ER-dependent signaling pathways. A number of cell-based and animal models have conclusively demonstrated that ERβ exhibits an antagonistic effect on ERα -mediated transcriptional activity. As has been observed in a number of other NR signaling paradigms, chromatin structure is an important regulatory factor of ER activity. Uncovering the mechanisms behind the reduced activity of ERβ on chromatin templates and the role the AF-1 domain plays in mediating this effect will be important in interpreting the functional roles of each ER subtype. Researchers are only beginning to decipher the intricate interplay among ERα , ERβ , and their respective isoforms, and although a large body of data suggests that the functional differences between the ER subtypes reside within the N-termini, continued efforts will undoubtedly uncover new modes of action in ER signaling networks. The improved design of ERα - and ERβ -selective ligands and identification of subtype-specific signaling pathways will be essential in planning novel therapeutic strategies for the treatment estrogen-dependent pathologies.
Acknowledgments
This work was supported by grants from the Swedish Cancer Fund
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Jan-Åke Gustafsson, MD, PhD , is Professor of Medical Nutrition and Director of the Center for Biotechnology at Novum, the South Campus of the Karolinska Institute; his main interest is nuclear receptor signaling. He is foreign associate of the US National Academy of Sciences. Address correspondence to J-ÅG. E-mail jan-ake.gustafsson{at}mednut.ki.se; fax: + 46-8-779 87 95

Jason Matthews, PhD , received his undergraduate degree from the University of Western Ontario in Canada before pursuing his doctoral studies in biochemistry and environmental toxicology at Michigan State University in East Lansing. He is currently a postdoctoral fellow in Prof. Gustafsson’s laboratory at the Karolinska Institute, Department of Biosciences in Huddinge, Sweden.

