Transgenic Analysis of Human Drug–Metabolizing Enzymes: Preclinical Drug Development and Toxicology
Abstract
The cytochromes P450 (CYPs) are drug-metabolizing enzymes (DMEs), and constitute a large family of monooxygenases, arising from multiple genes, capable of catalyzing an extraordinary range of biochemical reactions, from the synthesis of cholesterol, bile acids, and steroid hormones to the metabolism of drugs, prodrugs, and xenobiotics. The CYPs, along with other DMEs, remain the subject of intense scientific research not only to ascertain their functions in vivo, but also to better understand how the expression of these enzymes is regulated. Transgenic technologies are providing important animal models for studying how DMEs—including the particularities of human DMEs—provide an adaptive response to cellular stress, establishing relationships between polymorphisms and disease susceptibility, and investigating how CYPs and other DMEs determine the pharmacological action of therapeutic drugs1.
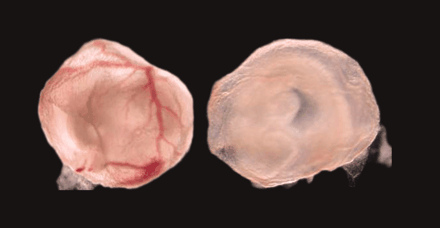
The lack of multiple cytochrome P450 activities in early development can be studied in a transgenic mouse. Most striking here
is the effect on blood vessel formation in the yolk sac of transgenic mice that lack cytochrome P450 reductase (right), relative
to wild type (left) 23).
Introduction
From biochemical studies and genome sequencing projects, we now know that a large number of genes, including those that encode the cytochromes P450 (CYPs), have evolved with the specific purpose of protecting cells from the deleterious effects of environmental chemicals. These genes are also pivotal in determining individual responses to therapeutic drugs. It is therefore of key importance to understand the roles of these genes, or families of genes, in determining cellular responses to chemical agents.
The multiplicity of drug-metabolizing enzymes (DMEs), including the phase I system (e.g., CYPs) and the phase II system (e.g., drug-conjugating enzymes such as UDP-glucuronosyltransferase and glutathione S -transferases), makes it extremely difficult to identify the in vivo function of any particular DME-encoding gene. In addition, there are significant interspecies differences in the functions of these genes, so that the relevance of animal models to human metabolism must be analyzed critically. This caveat extends to the use of transgenic technologies, the judicious use of which can provide a powerful means of establishing the in vivo functions of genes. In this short review, selected transgenic models will be described, and their potential uses in toxicology and drug development are discussed.
Generating Transgenic Animals
A transgenic animal may be defined as an animal species in which specific sequences of DNA have either been introduced or deleted from the genome. Currently, the major species of choice for such studies is the mouse. Although it is feasible to generate transgenic lines in a large number of other species, some of the technologies, such as the ability to delete specific genes from a genome, remain relatively undeveloped in species other than the mouse.
Two basic experimental approaches may be applied to generate transgenic animals (Figure 1A⇓). The first involves the random integration into a host genome of a specific sequence of DNA, in single or multiple copies, by injection into fertilized oocytes. This approach has the advantage of employing relatively straightforward, well-established methodologies, but can be problematic in allowing transgene constructs to integrate anywhere in the genome, so that unexpected positional influences on transcriptional regulation of the transgene (e.g., gene silencing) may occur. For example, a recent study of a sheep β -lactoglobulin transgene, maintained on a mixed CBA x C57BL/6 mouse background, shows that transgene expression can vary significantly among siblings (1) . This transgene was backcrossed for thirteen generations onto both parental backgrounds, resulting in significantly lower (although not silenced) expression in each parental background, relative to the mixed (CBA x C57BL/6) background; expression levels were restored by outcrossing. Interestingly, the authors observed “breeding problems” when backcrossing was attempted beyond the thirteenth generation in the CBA line, reminiscent of “genome policing,” a phenomenon described in plants, insects, and fish, whereby transgenes become silenced over the course of generations (2) .
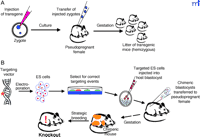
Transgenesis and gene knockout. A. Transgenesis is schematized as a process in which “foreign” DNA is injected into the pronucleus of a fertilized egg, which is then transferred to a pseudopregnant female. The resulting pups may contain one or more copies of the injected transgene, at an untargeted site (or sites) within the genome. B . A gene “knockout” is the deletion of a specific gene (or a whole family of genes) from a functional genome, so that the effect of gene deletion can be correlated to changes in drug metabolism, toxic responses, or susceptibility to carcinogenesis. At the present time, the technology for performing gene knockouts using embryonic stem (ES) cells is routinely available only for the mouse; however, in the near future, it is likely that other species, such as the rat, will also be amenable to this technology. In addition, it is possible to exchange genes by homologous recombination; for example, by deleting a particular gene or transcription factor and simultaneously substituting it with the same gene from another species. The organism produced by such a substitution (i.e., containing a transgene in place of an endogenous gene) is referred to as a “knockin” animal.
The second approach to generating transgenic animals is the targeted integration of DNA into the host genome using homologous recombination in embryonic stem (ES) cells (Figure 1B⇑). This technique allows the site of integration to be well-defined, so that it is possible to delete a specific gene or set of genes from the genome, introduce reporter constructs, or even to exchange pieces of genes between different species. The technology behind gene targeting is becoming increasingly sophisticated, and recently a new technique (i.e., “recombineering”) has been developed using phage-based homologous recombination in E. coli . This system makes it possible to engineer large DNA fragments in the form of bacterial or P1 artificial chromosomes (3) .
Equally important to the ability to study any given gene of interest per se, transgenic technology allows researchers to control the expression of their “favorite” gene product (e.g., at various developmental stages) and to correlate phenotypic changes to the “turning on and off” of transgenes. Such control of expression is especially crucial in studying genes the deletion of which are lethal in utero. Researchers have used a variety of protocols to construct transgenes that are amenable to conditional expression, and have indeed generated elegant stock mouse lines (through transgenesis) into which transgenes can be inserted and regulated at will. Examples of conditional transgene expression will be discussed below.
Some of the potential uses of transgenic animals for drug development and drug and chemical safety studies are shown in Table 1⇓. Table 2⇓ lists some of the commom problems, along with possible solutions, that may arise in the course of transgenesis.
Use of Transgenic Animals
Potential Problems with Transgenic Mouse Lines in Pharmacokinetics and Toxicology
Transgenic Animal Models in Drug Metabolism and Toxicology Studies
Cytochrome P450 Knockouts
Over the past decade or so, a number of CYP-encoding genes have been deleted in mice, as summarized in Table 3⇓. Generally, if the deleted CYP is essential to basal metabolism [e.g. CYP19 (aromatase) and CYP7A (cholesterol 7α -hydroxylase], then the phenotypic outcome is either embryonic mortality or perinatal morbidity, whereas if a gene is concerned with xenobiotic metabolism, its deletion may be inconsequential in the absence of a chemical challenge.
Cytochrome P450 Gene Deletions
The deletion of Cyp1a1 from the mouse genome produces no overt phenotype (4) . The Cyp1a1 knockout mice, however, are more robust, compared to the wild type, to the toxic effects of metabolizing benzo[a ]pyrene (BaP), although the retarded clearance of BaP results in significantly greater formation of BaP–DNA adducts (5) . Similarly, although Liang et al. report that the knockout of Cyp1a2 compromises neither viability nor fertility (6) , a subsequent report demonstrates a role for the enzyme in bilirubin degradation (7) as well as in protection against 2,3,7,8-tetrachlorodibenzo-p -dioxin–induced hepatotoxicity and uroporphyria (8) but not against acetaminophen hepatotoxicity (9) . Interestingly, Pineau et al., who also produced Cyp1a2 knockout mice, report such mice to be nonviable, suffering from severe respiratory distress shortly after birth, although there was incomplete penetrance of the phenotype (10) . As shown in Table 3⇑, the two groups not only used different targeting strategies to delete the Cyp1a2 gene, but also employed different genetic backgrounds.
Two other drug-metabolizing CYPs have been deleted in mice: CYP1B1 (11) and CYP2E1 (12) . Both knockout lines are viable, with no obvious phenotype, and only after chemical challenge [i.e., with dimethylbenz[a ]anthracene (DMBA) in the case of the Cyp1b1 knockouts and acetaminophen, chloroform, or carbon tetrachloride in the case of the Cyp2e1 knockouts] were differences found compared to wild-type mice. Double knockout mice, lacking both Cyp1a2 and Cyp2e1 , are significantly more resistant to acetaminophen toxicity than either of the single-knockout lines, and are fully viable after treatment with twice the dosage of acetaminophen found to be lethal to the wild type (13) .
In contrast, deletion of Cyp7a1 , encoding a key enzyme of cholesterol metabolism, results in almost complete neonatal lethality within two to three weeks (14) , although this phenotype is rescued through dietary supplementation with vitamins and cholic acid; the induction of an alternative bile acid biosynthetic pathway was reported through further study of the Cyp7a1 knockout mice (15) . Disruption of Cyp19 (i.e., aromatase), encoding a key enzyme in steroid hormone biosynthesis, results in viable neonates, although adult females are infertile (16, 17) . These results were reported independently by two research groups who used a similar targeting strategy and the same genetic background to generate their knockout mice. Two groups that knocked out Cyp26a1 , encoding an enzyme responsible for metabolism of retinoic acid and therefore crucial during embryonic development, used different targeting strategies, so that the knockout phenotype was described either as embryonic lethal (18) or lethal in the perinatal period (19) .
The multiplicity of genes that encode CYPs and the overlapping substrate specificity of the enzymes pose significant challenges when it comes to analyzing knockout experiments; the loss of a single enzyme may be partially or completely counterbalanced by one or more residual enzymes. The widespread distribution of Cyp genes throughout the genome makes their collective obliteration technically unfeasible. To circumvent this difficulty, the deletion of cytochrome P450 reductase (CPR), the sole electron donor for microsomal CYPs, was attempted (20, 21) . Because the CPR null mutation is embryonic lethal (22, 23) , the selective deletion of CPR from the liver alone is made possible by employing a (transgenically generated) mouse line that expresses Cre recombinase under the control of the rat albumin promoter (Box 1) (24) . Mice lacking hepatic CPR (i.e., HRN™ mice) are viable and fertile but completely devoid of liver CYP function; as a result, they are unable to metabolize testosterone, acetaminophen, and pentobarbital (26) . Such mice, in which an entire gene family has been inactivated as a consequence of deleting a single gene, will be of enormous use in delineating the specific role of hepatic P450s in drug metabolism and disposition. Interestingly, HRN™ mice, in contrast to Cyp7a single-knockout mice, display substantial alterations in lipid homeostasis, with levels of serum cholesterol and triglycerides lowered by 60–70% despite significantly elevated lipid content.
Conditional Gene Deletion
Essentially, conditional gene deletion means that the effect of the transgene or the genetic manipulation is silent until activated in a particular cell type or at a particular time. Cre recombinase is a bacteriophage enzyme that has the capacity to delete fragments of DNA in somatic cells if they are flanked by specific DNA recognition sequences called loxP sites (25) . When Cre recombinase activity is activated, either at a particular developmental stage or in a particular cell type in adults, genes flanked by loxP sites will be excised. This is a particularly powerful approach for studying the function of lethal genes during development; we have recently described this approach to study cytochrome P450 reductase (CPR) (26) . Cre recombinase has also been used in conjunction with the rat Cyp1a1 promoter in conditional transgenic systems; this promoter is highly activated in the presence of polycyclic aromatic hydrocarbons (27) . By linking Cre to the Cyp1a1 promoter, expression of the recombinase, along with Cre-catalyzed gene excision, becomes dependent on the presence of polycyclic aromatic hydrocarbons such as 3-methylcholanthrene or β -naphthoflavone (C.J. Henderson and C.R. Wolf, unpublished observations).
Beyond CYPs: Other Drug-Metabolizing Enzyme Knockouts
Several other DMEs, both Phase I and Phase II, have been inactivated by gene deletion, as is summarized in Table 4⇓. To date, the only report of a glutathione S -transferase knockout mouse, wherein both GstP genes were deleted, reports no overt phenotype (28, 29) . However, when tested in a two-stage skin carcinogenesis test, initiating tumorigenesis with DMBA and promoting tumor growth with 12-O -tetradecanoylphorbol-13-acetate, GstP null mice form skin papilloma at a significantly higher rate than wild-type mice, underlining the importance of this enzyme in protecting against chemical challenges (28) . Further characterization of this mouse model revealed the unexpected finding that mice lacking GstP are almost completely resistant to the hepatotoxic effects of acetaminophen. This resistance is surprising, given the evidence for the role of glutathione S -transferase P activity in detoxifying N -acetyl-p -benzoquinonimine, the toxic intermediate of acetaminophen (30) . Although still unclear, it appears that glutathione S -transferase P may play an important role in maintaining glutathione homeostasis, possibly through its recently reported ability to regulate c-Jun kinase (31) .
Other DME Gene Deletions
NAD(P)H quinone oxidoreductase 1 (NQO1) is a flavoprotein that catalyzes the two-electron reduction of quinones, and is induced as part of a coordinated cellular response to oxidative stress. Other enzymes that are induced in this response include the glutathione S -transferases, UDP -glucuronosyltransferase, epoxide hydrolase (EH), and glutamate-cysteine ligase (GCL). Deletion of NQO1 in mice causes no discernible developmental phenotype; however, NQO1 null mice are significantly more sensitive to the toxic effects of menadione (32) , develop myelogenous hyperplasia as a result of altered redox status, and show reduced rates of apoptosis in the bone marrow (33) .
EH is a phase I DME that imparts cellular protection by catalyzing the hydrolysis of a wide range of potentially toxic reactive epoxide intermediates. Both the soluble (sEH) and microsomal (mEH) forms of the enzyme have been inactivated by gene targeting. Although mEH null mice demonstrate no obvious phenotype, they are resistant to DMBA toxicity in a two-stage skin carcinogenesis model (34) , whereas deletion of sEH results in mice with aberrant renal arachidonic metabolism and, in the female, significantly lowered systolic blood pressure (35).
Glutathione peroxidase can be knocked out without causing developmental peculiarities relative to the wild-type mouse (36, 37) , although its deletion renders mice significantly more susceptible to the toxic effects of Diquat, an herbicide that reacts with molecular oxygen to generate superoxide anions and hydrogen peroxide. Glutathione peroxidase thus appears to subserve an antioxidant function in vivo (38, 39) .
Although not normally defined as a DME, glutamate-cysteine ligase is a key enzyme in the biosynthesis of glutathione, which in turn plays a central role in cellular protection against endogenous and exogenous electrophiles. GCL is dimeric, containing a catalytic and a regulatory subunit. Mice lacking the regulatory subunit of GCL display significantly lower levels of glutathione in several tissues, including liver and lung (40) . Knockout mice lacking the regulatory subunit have therefore been proposed as a model for chronic glutathione depletion as occurs in disease states and under environmental oxidative stress.
Regulators of Drug-Metabolizing Activities As Knockout Targets
Aryl Hydrocarbon Receptor
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates the toxic and carcinogenic effects of polycyclic aromatic hydrocarbons. AhR functions in a heterodimeric association with the AhR nuclear translocator (ARNT); both proteins belong to the family of basic helix-loop-helix/PAS transcription factors. Three research groups have independently deleted AhR, and the results of these studies illustrate clearly some of the potential problems that can arise in transgenic manipulation of animals. Of the three groups, two used a targeting strategy involving disruption of exon 2 (49, 50) , and both reported similar phenotypes of viable, fertile mice, albeit with initially lower growth rates and a number of hepatic defects. Additionally, the construct used by Mimura et al. also contained a lacZ reporter gene, enabling the authors to study the expression pattern of the AhR gene during development and in the postnatal period (41) . The third group disrupted exon 1 of the AhR-encoding gene and found a more complex phenotype: 50% of these AhR null pups die at birth and 50% survive to adulthood. The adults are fertile, but display significantly smaller liver weights, extensive hepatic fibrosis and cholangitis, and decreased lymphocyte populations in the spleen and lymph nodes (42) . Because all three AhR knockout mouse lines were on a mixed 129xC57BL/6 genetic background, it seems likely that the disparate targeting strategies may underlie the observed phenotypic differences. Proposed explanations for the different phenotypes include differential exposure to environmental chemicals, pathogens, and other stressors (43, 44) .
ARNT has also been deleted by independent research groups. Two groups performed a direct knockout, both eliciting embryonic lethality but observing distinct spectrums of defects. Whereas Maltepe et al. (45) drew the conclusion that Arnt plays an essential role in the promotion of angiogenesis and vascularization in both the yolk sac and developing embryo, Kozak and colleagues found yolk sac circulation to be essentially normal, and ascribed the death of Arnt null embryos to placental insufficiency (46) . In terms of targeting strategies, both groups disrupted the same region of the Arnt gene, and their mouse lines had the same genetic background (129/SvxC57BL/6); however, Kozak et al. note that the 129 ES cells used in their study were distinct from those of Maltepe et al. in terms of genetic origin.
A third research group, acknowledging that the deletion of ARNT elicits embryonic lethality, flanked part of the Arnt gene with loxP sites and introduced a Cre transgene driven by the interferon-γ promoter (47) . Activation of the Cre transgene, causing inactivation of Arnt , results in mice that no longer manifest the AhR-mediated inducibility of genes in the liver. Moreover, despite an 80%-reduction of Arnt expression in the lung, induction of CYP1A1 by AhR was found to be maximal, indicating that Arnt expression is not rate-limiting for AhR function.
Nrf2: A Transcription Factor in Antioxidant Response
Nuclear factor-erythroid 2–Related Factor 2 (Nrf2), a member of the basic leucine zipper family of transcription factors, binds to the antioxidant response element found in the regulatory regions of a number of genes, including glutathione S -transferase and NQO1, in response to chemicals such as butylated hydroxyanisole (48) . By deleting Nrf2, Chan et al. found that Nrf2 is not essential for development (49) , a finding that was confirmed independently by Itoh and colleagues (50) .
Nrf2 null mice have been used in a series of studies to demonstrate the importance of this transcription factor in regulating the expression of genes that protect against oxidative stress. Mice lacking Nrf2 are very sensitive to acetaminophen hepatotoxicity (51, 52) as well as to pulmonary damage caused by either butylated hydroxytoluene (53) or hyperoxic conditions (54) . Recently, Noda et al. (55) reported the generation of a compound double null (AhR and Nrf2) mouse, which lack the ability to respond to chemical inducers that signal through the pathways regulated by these transcription factors. It is worth noting that the genetic background of these mice is more heterogeneous (Table 5⇓) as a result of crossing individual knockout mice, and that perinatal mortality was approximately 60% for the double null mice.
Receptor and Transcription Factor Deletions
PPARs
Peroxisome proliferator–activated receptors (PPARs) are lipid-activated transcription factors that belong to the nuclear receptor superfamily. Three types of PPAR have been identified: PPARα appears to mediate a set of responses induced by a wide array of chemicals collectively known as peroxisome proliferators; PPARγ is involved in adipogenesis and differentiation; and PPARδ (β ) seems to be involved in lipid metabolism (56) .
PPARα knockout mice have no overt phenotype, but all responsiveness to peroxisome proliferators is abolished (57) . In addition, these mice are protected against the development of insulin resistance following exposure to a high-fat diet (58) . The production of mice devoid of PPARδ (β ) was initially difficult, and only against an enhanced C57BL/6 genetic background did the knockout yield homozygous null pups born in Mendelian proportions (59) . These mice are smaller, both in utero and postpartum, than their wild-type counterparts, implying a developmental role for PPARδ (β ). PPARδ (β ) null mice also have smaller gonadal adipose stores, reduced myelination in the brain, and an increased hyperplastic response to the application of phorbol ester to the skin (59) . Double knockout mice, devoid of both PPARα and PPARδ (β ), show impaired wound healing (60) .
Knockout of PPARγ activity is embryonic lethal, resulting in placental insufficiency leading to cardiac deficiency (61) , although a term mouse can be produced through supplementation of embryos with wild-type placenta. Nevertheless, the placentally rescued phenotype manifests severe health problems after a few days involving an almost complete lack of adipose tissue and bleeding in the intestine and brain (61) . A conditional PPARγ deletion in macrophages has been reported to result in altered cholesterol homeostasis through changes in cellular efflux and plasma transport (62) .
Key transcription factors involved in the regulation of expression of DMEs that have been deleted by gene targeting appear in Table 5⇑.
Humanization of Mice for Drug Development Models
One of the major goals of knockout and transgenic mice is to understand the function and regulation of human DMEs. The replacement of a given rodent gene by its human analog requires significant homology between the two. For example, the pregnane X receptor (PXR) found in rodents and the human homolog steroid and xenobiotic receptor (SXR) have been postulated as candidate xenosensors in the regulation of CYP3A-encoding genes; indeed, deletion of PXR in the mouse results in the ablation of CYP3A induction in response to xenobiotics (63) . Expression of the human SXR transgene on the PXR null background restores CYP3A inducibility, although only in response to human-specific inducers such as rifampicin (63) . This system has great potential for the development of new pharmaceuticals, because CYP3A4 is involved in the metabolism of as many as 50% of currently prescribed drugs. By screening novel test compounds early in the development process for their ability to induce CYP3A, it may be possible to avoid a significant number of undesirable drug–drug interactions. CYP3A4 is also abundantly expressed in the intestine, and Granvil et al. report the development of a transgenic mouse line in which this enzyme is expressed with a profile similar to that found in humans (64) .
CYP2D6 is responsible for the metabolism of more than 100 commonly used drugs in humans (65–67) . Due to interspecies differences in the metabolism of CYP2D6 substrates, the use of animal models to predict drug metabolism in humans may be limited (68) . To circumvent metabolic inconsistencies between species, Corchero and colleagues generated a mouse expressing a humanized CYP2D6-encoding gene and thereby promoting the 4-hydroxylation of debrisoquine, a reaction typical of the human CYP2D6 enzyme (69) . Moreover, the addition of the human HNF4α transgene satisfies the need for this transcription factor in the regulation of transgenic CYP2D6 expression (69) .
The relative dearth of reported humanized mice underscores not only the technical difficulties in generating such transgenic lines, but also the problems in creating meaningful models, where the spatiotemporal expression pattern and regulation of the transgene mirrors that of the gene in its “normal” environment.
Transgenic Reporter Systems
Many of the transgenic and knockout systems described above have also incorporated a reporter gene in order to gain further information concerning the regulation and expression pattern of the gene of interest. Most often, the reporter is lacZ , but increasingly, green fluorescent protein or luciferase is utilized. Often, although deletion of the desired gene is successful, the reporter system does not work, for reasons that are not always clear but probably involve mutation of the targeting construct and/or unappreciated regulatory idiosyncrasies of both the donor transgene construct and host transcription machinery. Campbell et al. were able to circumvent problems of this latter sort by employing a large fragment (8.5 kb) of the rat Cyp1a1 promoter in order to include potentially distant regulatory elements important in controlling gene expression (27) ; in so doing, expression of a lacZ transgene became tightly regulated and highly effective as a reporter. Properly implemented, reporter systems provide a much higher level of sensitivity and definition in studying protein expression than can be obtained using by other methods, such as immunostaining. Reporter systems are also unrivaled in providing real-time information on cellular changes within living animals.
A number of in vivo reporter systems have been devised around the inducible CYP1A1 gene promoter, due to the fact that this system exists in essentially only two states, either on or off, depending on the presence or absence of a polycyclic aromatic hydrocarbon. Jones et al. (70) made use of the mouse Cyp1a1 promoter linked to a chloroamphenicol acetyltransferase reporter and found low basal expression levels in several tissues. Upon treatment with polycyclic aromatic hydrocarbons, however, reporter activity was found to be highly induced (> 10,000-fold in liver). A similar strategy was adopted by Smith and colleagues (71), who employed a short region of the Cyp1a1 promoter linked to the human gene that encodes apolipoprotein E. Multiple independent transgenic lines displayed low constitutive APOE expression that was greatly elevated by induction with β -naphthoflavone. The β -naphthoflavone-inducible APOE transgene was further bred onto a hypercholesterolemic ApoE -null background. The resulting hypercholeserolemic mice could then be treated with β -naphthoflavone, which significantly lowered blood cholesterol, demonstrating a potential therapeutic strategy for human genetic anomalies.
Conclusion
The application of transgenesis to toxicology, preclinical drug development, and drug safety studies has enormous potential to increase our understanding of how chemical agents interact with cells, provided that expression data are interpreted with care. Particular vigilance should be exercised with regard to genetic background, because the presence of “modifier” genes in different backgrounds may alter the phenotype(s) observed (72). It is important that this potential effect is taken into consideration, and it is generally prudent to perform backcrossing onto a defined genetic background (73). The use and application of in vivo transgenic models can be costly and time-consuming, although these drawbacks are being continually mitigated. The power of these approaches is alluring, promising to reveal the mechanisms by which humans can respond to environmental agents.
Acknowledgments
The work of colleagues within the Biomedical Research Centre is gratefully acknowledged. Work in the Molecular Pharmacology Unit is funded by Cancer Research UK.CRW is a Director, cofounder, and shareholder of the Dundee biotech company CXR Biosciences Ltd. CJH is a consultant to CXR Biosciences.
Footnotes
-
↵1 This review is part of a series on cytochrome P450 enzymes.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

C. Roland Wolf, PhD, (upper photo) is Professor and Director of the Biomedical Research Centre at the University of Dundee. Colin J. Henderson, PhD, (lower photo) is Staff Scientist and Honorary Lecturer at the Biomedical Research Centre. Address correspondence to CJH. E-mail colin.henderson{at}cancer.org.uk; fax +44 1382-669993.

