LIPID Arrays: New Tools in the Understanding of Membrane Dynamics and Lipid Signaling
- Department of Pharmacology and the Vanderbilt Institute of Chemical Biology, Vanderbilt University School of Medicine, Nashville, TN 37232
- Address correspondence to HAB. E-mail alex.brown{at}vanderbilt.edu; fax 615-343-6532.
Abstract
Phospholipids are the structural building blocks of the membrane bilayer, which retains and regulates intra-cellular content. In addition to creating a protective barrier around the cell, lipids modulate membrane trafficking and are themselves precursors of important intracellular signaling molecules. Identification and quantification of these molecular species is essential for a more complete understanding of cell signaling pathways, and more reliable and sensitive methods are needed for determining membrane phospholipid content. Recent improvements in electrospray ionization mass spectrometry have made possible the direct identification of more than 400 phospholipid species from biological extracts of a single cell type. Changes in the cellular concentration of diverse lipids can be determined by analysis of the mass spectra by statistical algorithms. In the future, lipid arrays will be integrated with other high-throughput profiling technologies, and computational lipidomics will expand our understanding of the molecular basis of cellular processes and diseases.
Introduction
Life science has entered a new era, in which the wealth of genomic, proteomic, and small-molecule data can be overwhelming. Although terabytes of data will never replace the essential requirement for hypothesis-driven questions, new technologies are allowing investigators to more comprehensively identify the missing pieces of complex biological puzzles prior to assembly. As a result, the need for high-quality bioinformatics to manage and integrate experimental data becomes imperative.
Increasingly, lipids are recognized as important participants in the regulation and control of cellular function and disease, intimately involved in signal transduction processes (1, 2). The diversity in their chemical structure presents a challenge for establishing practical methods to generate and manage high volumes of complex data that translate into a snapshot of cellular lipid changes. A truly comprehensive “lipidomics” approach must incorporate multiple separation and identification techniques, manifesting sufficient sensitivity to distinguish closely related metabolites while remaining robust enough to cope with the wide variety of heterogeneous classes of cellular lipid species. Electrospray ionization mass spectrometry (ESI-MS), with its extraordinary sensitivity and its capacity for high throughput, has become the technique of choice for the analyses of multicomponent mixtures of lipids from biological samples.
Phospholipids in Signaling
Lipids had traditionally been viewed as passive components of the cell membrane. Work over the last forty years from pioneering researchers, including Kennedy, Samuelsson, Berridge, and Mitchell, has revealed that lipids are inextricably linked to such fundamental physiological processes as bioenergetics, cellular recognition, and signal transduction across the cell membrane. When the cell responds to external signals, a cascade of intracellular enzymatic activation ensues that can elicit changes in the composition of membrane lipids. One major group of cellular lipids involved in signal transduction comprises the glycerophospholipids (henceforth referred to as phospholipids), whose general structure is illustrated in Figure 1⇓.

Glycerophospholipid structure. Typical glycerophospholipids consist of a glycerol backbone, two acyl moieties, and a phospho(di)ester headgroup. Arrows indicate the sites of phospholipase-mediated hydrolysis. (PLA, phospholipase A; PLC, phospholipase C; PLD, phospholipase D; PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PIP, PI monophosphate; PIP2, PI bisphosphate; PIP3, PI trisphosphate.)
Phospholipid molecules consist of a glycerol backbone esterified, at the sn-1 and sn-2 positions, by carboxylic acid residues. The sn-3 position is occupied by a phosphate moiety, which is connected via a phosphoester bond to the so-called “headgroup;” phospholipids are grouped into classes according to the identity of the headgroup. The major classes of phospholipids found in the membrane of mammalian cells include phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidylglycerol (PG). The variety in length and the degree of unsaturation found in the acyl chains create a large diversity of molecular species within a class. There can be additional variations within a given phospholipid class: in place of ester linkages, vinyl ethers (O-CH=CH-R) or saturated ethers (O-CH2CH2R) may modify the glycerol backbone at the sn-1 position, creating the plasmenyl or plasmanyl phospholipids, respectively.
Phospholipid metabolism is regulated through both G protein– and tyrosine kinase–mediated enzymes, including the phospholipases C, D, A1 and A2 (PLC, PLD, PLA1 and PLA2) as well as several lipid kinases and phosphatases. These enzymes convert lipids into bioactive signaling molecules, such as phosphatidic acid (PA), diacylglycerol (DAG), and polyphosphoinositides (3–5). As illustrated in Figure 2⇓, lipid signaling mechanisms form a complex web of interrelated pathways. Consider, as an example, the multiple routes for the generation of the lipid-derived second messenger DAG, which may be formed via an agonist-mediated G protein–coupled receptor (GPCR) through the concerted action of Gαq and PLCβ, or via a PLCγ isoenzyme acting downstream of a cell-surface tyrosine kinase. Alternatively, DAG can be formed from the turnover of PA by lipid phosphate phosphatase (LPP). Many of the enzymes that act on phospholipids are specific for the acyl composition of the lipid as well as for the headgroup. For example, the epsilon isoform of mammalian diacylglycerol kinase (DGKε) strongly prefers substrates with an arachidonoyl group at the sn-2 position. In addition to its headgroup and acyl moieties at sn-1 and sn-2, subcellular localization and signaling state of the cell will determine the fate of a particular bioactive lipid at any time. These aspects of phospholipid turnover have been explored; however, the effects of phospholipid signaling on subsequent membrane remodeling are only beginning to be determined. A deeper understanding of the downstream changes in lipid composition following receptor-activated signaling is essential given that the alterations have profound effects on cellular function, intricately modulating the outcomes of diverse receptor activation at the cell surface. For example, whereas multiple receptors on a given cell type share the property of activating some isoform of PLC, there is likely to be great diversity between different receptor types in terms of the ensuing signaling events downstream of catalysis by PLC. A detailed examination of cellular phospholipid species that change as a result of single or dual ligand additions is being conducted as part of our studies for the Alliance for Cell Signaling (http://www.signaling-gateway.org/). Among our goals, we seek to determine whether distinct phospholipid species are deployed by distinct isoforms of PLC, PLD, or PLA2, thereby propagating distinct signaling pathways. A diversity of signaling pathways may be caused by the differential activation of enzyme isoforms downstream of distinct GPCRs or by some combined activation of cell surface receptor pathways. Similarly, determinations of the complementary lipid profiles that accompany generation of prostaglandins (e.g., PGE2; see Figure 2⇓) will help us to more fully understand the contributions of specific lipid metabolic reactions within the cell membrane to networks of signaling pathways. For example, as illustrated in Figure 2⇓, GPCR activation often leads to the stimulation of PI 3-kinase which, in turn, stimulates guanine nucleotide exchange factors (GEFs), such as those required to activate the monomeric GTPase Rho. In parallel, these receptors also lead, via Gαq PLCβ to the generation of DAG and inositol 1,4,5-trisphosphate (IP3). The production of DAG and mobilization of calcium activate certain classical isoforms of protein kinase C (PKC). This simultaneous activation of PKC, formation of GTP-bound (i.e., activated) Rho, and increased availability of multiphosphorylated forms of PI appear to converge to permit the activation of phospholipase D (PLD). The fate of these signaling lipids and the contextual lipid changes that follow are largely unknown.
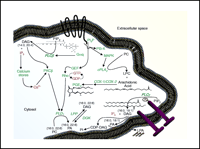
Lipid signaling pathways in mammalian cells. Some of the richness and complexity of lipid signaling is conveyed by the conventional designations shown above: DAG, for instance, is not a single species, but rather a variety of diglycerides with varying acyl chains. Distinct cell signaling pathways may mediate the generation of specific lipid products through the use of differential isoenzymes (e.g., PLCβvs PLCγ). Similarly, DGKεhas a preference for 20:4 DAG as a substrate. Lipidomic approaches identify distinct lipid species, such as specific acyl species of DAG, according to their individual roles in signaling. In other words, not all DAGs may be created equally nor have the same effects on the cell. Further elucidation of the signaling pathways shown above into discrete reaction mechanisms will facilitate understanding the roles of lipids in cellular processes. Proteins are indicated in green text, lipid species in black text, and other small molecules in red text; a GPCR is shown as a black band with seven transmembrane domains; a pair of nail-like geometries represents an active receptor tyrosine kinase (lower right). (PC, phosphatidylcholine; PI, phosphatidylinositol; PA, phosphatidic acid; DAG, diacylglycerol; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; IP3, inositol 1,4,5-trisphosphate; PIP2, phosphatidylinositol bisphosphate; PIP3, phosphatidylinositol trisphosphate; PLC, phospholipase C; PLD, phospholipase D; PLA, phospholipase A; LPP, lipid phosphate phosphatase; DGK, diacylglycerol kinase; GEF, guanine nucleotide exchange factor; PKC, protein kinase C; PI3-K, phosphatidylinositol 3-kinase; MAPK, mitogen activated protein kinase; COX, cyclooxygenase; PGE2, prostaglandin E2; CDP-DAG, cytidine diphosphate-diacylglycerol.)
Phosphatidic acid, a product of regulated hydrolysis of PC by PLD, has been associated with a number of cellular processes, such as secretion, endocytosis, and cell cycle progression, as well as activation of protein and sphingosine kinases (6). Lysophosphatidic acid (LPA) can be generated via several signaling pathways, including hydrolysis of PA by PLA2. Alternatively, lysophospholipase D (lysoPLD) can produce LPA from lysophosphatidylcholine or from a number of other metabolic routes. LPA has been linked to differentiation, growth promotion, motility, and survival in various cell lines, and may thus play important roles in physiological and pathophysiological processes including inflammation, angiogenesis, and carcinogenesis (7–10).
Phosphoinositides (phosphorylated phosphatidylinositols) are another class of phospholipids that are involved in important cell signaling pathways (11). Phosphatidylinositol-3,4-bisphosphate (PIP2, lipid “second messenger”) binds to and activates Akt, which is critical to numerous biological processes, including programmed cell death (12, 13). The implications of phospholipids in a variety of signal transduction and enzymatic pathways that contribute to human disease are only beginning to be realized (14–16). A more complete picture of lipid metabolism and regulation will emerge as new analytical techniques overcome measurement limitations related to the diversity, low abundance, and rapid inter-change reactions of lipid species.
Analytical Techniques for Phospholipid Analysis
The conceptualization of specific phospholipid constituents in terms of membrane transport, signal transduction, and other changes in membrane processes must be predicated on a consideration of the exact lipid composition of the given cell type. Meaningful physiological integration into such processes will require the elucidation of the chemical make-up and quantification of each phospholipid species. Conventional methods for the structural characterization of membrane phospholipids are typically multistep procedures (17). Classical liquid-liquid extraction of cellular material readily results in heterogeneous mixtures of phospholipids, which can be further refined by the selective use of organic solvents. The next step is a class separation, generally relying on analysis by thin-layer chromatography, high-performance liquid chromatography (HPLC), and gas chromatography, any of which can necessitate prior derivatization as well as the availability of known standards (18–21). This conventional approach is time-consuming, requiring relatively large sample volumes for accurate separation, and lacks the sensitivity required for quantifying minor components, which often include the crucial signaling species.
Characterization of phospholipid species by mass spectrometry (MS) had long been hindered by their propensity to undergo fragmentation during analysis, but has recently become possible in large part because of dramatic technical advances. Since the original studies of J.J. Thomson (22) and F.W. Aston (23) at the beginning of the last century, MS has in fact evolved into a driving force that is revolutionizing the ability to detect and analyze a wide variety of biological molecules. The combination of sensitivity, specificity, selectivity, and speed makes MS an ideal technique for analysis of phospholipids.
Field desorption was the first MS method used to analyze intact phospholipids (24). With the development of fast atom bombardment MS (FAB-MS), natural phospholipids were first observed to generate abundant molecular (i.e., unfragmented) ions along with fragment ions (25, 26), which provided some structural information and revealed the great potential of tandem MS MS/MS; i.e., analysis of molecular and fragment ions generated therefrom) for the structural characterization of lipids. Nevertheless, the challenges associated with the thermolability and nonvolatility of lipids precluded the routine use of these methods in phospholipid analysis until the advent of “soft” ionization techniques such as matrix-assisted laser desorption/ionization (MALDI) (27) and electrospray ionization (ESI) (28, 29), innovations that allow for the direct analysis of intact, polar, thermally labile and highly nonvolatile biomolecules. “Soft” ionization does not cause extensive fragmentation, so that comprehensive detection of an entire range of phospholipids within a complex mixture can be correlated to experimental conditions or disease state. Various ESI-MS methods have been developed for analysis of different classes, subclasses, and individual lipid species from biological extracts. Comprehensive reviews of the methods and their application have recently been published (30–33). The major advantages of ESI-MS are high accuracy, sensitivity, reproducibility, and the applicability of the technique to complex phospholipid solutions without prior derivatization.
The development and application of ESI tandem MS (ESI-MS/MS) to lipid analysis over the last ten years has reflected contributions from many research groups (see, for examples, 34–37). The automation of sample injection and fast analysis time enable high-throughput analyses. The development of computational analysis of MS data has also made the assessment of phospholipid composition more feasible. The changes in cellular concentrations of numerous lipid species can be monitored simultaneously in response to a biological stimulus. The conversion of such data into conceptually accessible lipid arrays has been described in detail (38, 39) and will be reviewed below. Detailed descriptions of the methods of cell stimulation, extraction, and sample preparation for MS analysis are provided elsewhere (39).
Application of ESI-MS for Phospholipid Analysis
ESI-MS was initially developed by Fenn and colleagues (28) and relies on the creation of gaseous ions from polar, mostly nonvolatile, molecules such as lipids. The principle of ionization involves a sample in solution that is passed through a very thin capillary at a slow rate (1–300 μl/min). The application of a strong electric field generates significant charge at the end of the capillary that produces a fine spray of highly charged droplets. These droplets then pass through a heated inert gas for desolvation prior to mass spectral analysis of individual ionic species (Figure 3⇓).
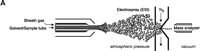
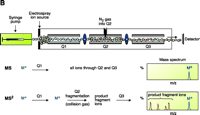
Electrospray ionization and triple quadrupole mass spectrometer. ESI-MS is unsurpassed for complete analysis of phospholipid mixtures. It has been successfully used for quantitative measurements of phospholipids from biological samples (40–43). The “soft” electrospray ionization causes little or no fragmentation and hence virtually all phospholipid species can be detected as molecular ion species (denoted as M*), when analyzed in a single-stage mode using either Q1 or Q3 quadrupoles operating as mass analyzers. The choice of polarity is determined by the possible charge state of the phospholipid class in solution. All zwitterionic phospholipids (PC, LPC, SM, and PE) can ionize in both positive and negative ion modes. With the exception of PE, they are more efficiently analyzed in positive mode. Because of their negative charge at neutral pH, anionic phospholipids (PI, PA, PG, and PS) produce primarily [M-H]- (deprotonated molecule retaining a negative charge) as the molecular ion peak and are easily detected in negative ion mode. Thus, two sets of mass spectra are obtained for each sample—one in positive and one in negative ionization mode, with all detected phospholipid species presented as peaks at their specific m/z value. The identification and structural information about a particular peak is acquired by tandem MS (44, 45). The relevant ion is subjected to collision-induced dissociation (CID) by interaction with a collision gas. A fragmentation spectrum (product, or “daughter” spectrum) is generated as a result, revealing structural information (acyl chain or head group chemistry, depending on the ionization mode). On that basis, “fragmentation libraries” can be created for the phospholipid species composition of any cell type (38).
The characterization of individual phospholipid species from an unprocessed (i.e., compounds that were not derivatized) total lipid extract by ESI-MS/MS is based on the ability of any given phospholipid class to acquire charge (positive or negative) when in solution under the capillary source high energy. Lipids that do not possess inherently ionizable functions (e.g., diacylglycerols) can also be ionized under the conditions of ESI-MS, and as long as there is a sufficient dipole potential for interaction with small counter ions, these species can be analyzed. Thus, under suitable conditions of sample preparation, all molecular species that exist among cellular lipid classes can be detected in a single run of a total lipid extract. They are identified on the basis of their characteristic m/z (mass-to-charge) value and their structure is confirmed by fragmentation analysis (tandem mass spectrometry, MS/MS). Typically, cellular lipid profiles under two experimental conditions can be monitored over a given time course. With the advancement in MS instrumentation, data analysis rather than data acquisition has become the rate-limiting step.
Mathematical Analysis of Mass Spectrometry Data and Generation of Lipid Arrays
Having described advances in MS technology and its application to cell signaling, we now address a novel approach to the computational analysis of global pattern changes in membrane lipid composition. When using a triple quadrupole mass spectrometer in full-scan mode, the user typically selects an upper and lower m/z value for the scanning range. The instrument software divides this area into equal partitions where ion intensities are recorded during sample delivery. Thus, using a typical m/z range for phospholipids of 400 to 1200 with a gradation of 0.10, a single experiment would collect 8000 data points over the given m/z range. When considering multiple time points and several experimental replicates, the amount of data collected is massive, requiring computational tools for managing and compressing it into a conceptually useful form. The combination of several statistical ideas allows the examination of such MS output in an arrayed organization that reflects the form and presentation of gene expression microarrays. The goal of this analysis is the rapid, objective identification of statistically significant differences in mass spectra between two biological conditions. We have developed computer algorithms for this analysis using the S-Plus Version 3.3 for Windows programming suite (StatSci division of Mathsoft Inc., Seattle, Washington). The mathematical analysis relies on the two key concepts outlined below.
Data Normalization
The functions relating the observed ion intensities to the relative amounts of the components in a complex biological extract are often complex and difficult to identify. Ionization efficiencies vary from compound to compound and are further dependent on the particular environment in which samples are analyzed. To over-come these complexities, overall spectral patterns can be statistically “normalized” so that data sets can be compared. Several types of normalization schemes have been included in our software algorithms. The more robust transformations across experimental replicates have been based either on conversions using standard statistical units or on signal ranks. In the first instance, the ion intensities are replaced with the central limit theorem type transformation: xi → (xi−x)/σx, where x is the mean of the intensities and σx their standard deviation. In converting to standard units, a signal with intensity identical to the average for the given data set would be expressed as zero, and any signal with a lower-than-average intensity would be expressed as a negative value. The second type of transformation maps each intensity to its rank as compared with the other measurements in the data set (with a rule for identical intensities). Thus, if 8,000 data points are transformed, they are uniquely assigned an integer value from 1 to 8,000. Once a normalization scheme is selected, the intensities are transformed and these values are utilized in the second part of the analysis.
Shewhart Control Charts
Shewhart control charts are mathematical devices commonly used in statistical process control and are ideal for identifying signal differences between two biological conditions. The original usage of these charts was in the monitoring of industrial processes, where complex interactions among equipment, procedures, and environmental conditions required statistical tools to separate common cause from assignable cause variation. The construction of a control chart involves drawing samples from the process under study at selected time points and computing a statistic of interest, such as the mean. These values are then plotted as a time series along with an upper and lower control limit, which are calculated from the data. These limits represent the expected variability in the plotted statistic, assuming the process remains stable. A process is labeled as “in-control” if it exhibits only random variation with respect to the time grouping. This is defined as meaning all sample statistics (not individual measurements) are within the control limits and no non-random patterns are present. In analyzing the MS of phospholipid samples from an experimental set of conditions, m/z values where the transformed signal is in-control over the time course in the basal condition represent molecules in which the components of measurement variance are stable and the signal is not subject to time-based drift (i.e., in-control lipid species are at steady state in the absence of external stimulation).
Computational Analysis
The analysis programs used in the construction of the lipid arrays utilize the concepts discussed above. Data files from the mass spectrometer are first smoothed to remove the shoulder effect from the various peaks in the spectrum. After smoothing, each data set is normalized as described above and parsed for peak locations. Peaks are identified by the software as “Low-High-Low” patterns appearing in the transformed ion intensities. These peaks are then collected in a data table where their final locations (m/z ratio) are the intra-group averages of the individual locations.
In the next phase of the analysis, a Shewhart control chart for the means of normalized signals is constructed for every peak identified in the basal condition. Each chart is tested to determine if the signal is in-control. At each m/z value where the basal signal was found to be stable over the time course, the control limits are recorded for comparison with the stimulated condition. Using these limits, the algorithm then examines the means of the transformed data from the stimulated condition for non-random variation, including parsing for means beyond the control limits as well as searching for non-random patterns. If the basal data do not display time-based statistical control, extending the control limits for comparison with the stimulated data would be inappropriate. In this event, a two-sample t-test is performed at each of the time points to determine if differences exist in the means of the transformed data between the two conditions.
Generation of Lipid Arrays
After the primary computational analysis, color-coded lipid arrays are constructed to highlight changes in lipid species as a result of the biological conditions (i.e., stimulation of a G protein–coupled receptor or progression of a disease process). The vertical axis of the two-dimensional arrays represents m/z values (i.e., individual lipid species); the time course is represented on the horizontal axis. Lipid species that decrease in concentration over time as determined by a statistically significant decrease in normalized peak intensity are indicated in blue and assigned a numeric value of −1, whereas increases are colored red and scored as +1. Statistically stable combinations (i.e., no significant change in peak intensity) are scored with a 0 and are colored green. An excerpt of a lipid array is depicted in Figure 4⇓.
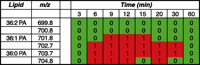
Excerpt of a lipid array displaying peaks in them/z range of 699.8 to 704.8 at 8 time points. Analysis using CID MS/MS determined that this area contains phosphatidic acid (PA) species with 36 carbons and three different double-bond configurations. Two of the species are increasing in intensity at several of the time points between 6 and 30 minutes following stimulation of B6A4C1 mast cells with the calcium ionophore A23187.
The goal of the lipid arrays is to winnow out significant differences in lipid profiles between two contrasting biological conditions in the presence of noise. In a typical eukaryotic lipid extract, each instrument mode will produce 700–900 peaks, of which 100–200 can be routinely matched to specific lipid species by CID MS/MS. Performing this number of statistical tests creates increased opportunity for false positives, similar to the problems inherent to genomic and proteomic approaches. One remedy is to conduct multiple repetitions of each experiment and to run replicate samples per time point, so that data from multiple arrays can be compiled. This repetition allows for the analysis of inter-experimental variation, which aids in the overall validation of conclusions. Once significant global changes in lipid profiles are identified, additional methods, such as inclusion of an internal standard, can be used to measure quantitative changes in a specific subset of the cellular lipome.
Demonstration of Lipidomics Technology
Mast Cell Degranulation: Cell Biology
Mast cells are involved in allergic inflammation responses, where-by they undergo “degranulation,” in an antigen- and Ca2+-dependent process, to release bioactive molecules, including phospholipids. Experimentally, degranulation can be monitored by assaying for the release of β-hexosaminidase, and as seen in Figure 5⇓, both the antigen and the Ca2+ dependency of degranulation can be assessed. The fusion between secretory vesicles (i.e., “granules”) and the plasma membrane, the final stage of exocytosis, can be further resolved into discrete steps. We have shown, for example, that the addition of exogenous phospholipases to permeabilized RBL-2H3 mast cells results in the production of bioactive lipids that trigger the release of granule contents through the plasma membrane, and that these particular phospholipase-dependent steps occur independently of cytosolic factors (37, 46).
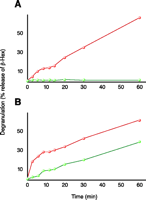
Degranulation in mast cells. Comparison between RBL-2H3 (red) and B6A4C1 (green) cells treated with antigen (A) and Ca2+ ionophore A23187 (B). The B6A4C1 cells have been shown to be defective in antigen-mediated degranulation while competent in calcium ionophore-mediated degranulation.
The redistribution of membrane phospholipids that occurs during degranulation is an additional aspect that should be amenable to study, leading to fuller insight into the regulation of secretory events. To analyze the phospholipid changes that underlie exocytosis more comprehensively, we measured lipid changes during antigen receptor–(FcεRI)–mediated degranulation in mast cells and compared them with changes induced by the Ca2+ ionophore A23187. We further studied two cell types: the well-characterized RBL-2H3 mast cell line and the B6A4C1 mutant mast cell line. As described above, lipid profiles of both cell types were monitored without treatment to establish in-control basal states (see 46).
The B6A4C1 cell line, derived from the RBL-2H3 mastocytoma line, is defective in FCεRI-coupled secretion, receptor-dependent calcium mobilization, ganglioside transport, and stimulated phospholipase activities (47, 48). Although defective in the antigen-stimulated reaction, B6A4C1 cells degranulate in response to the Ca2+ ionophore A23187 (Figure 5⇑). The changes in cell membrane phospholipid composition that result from physiological stimulation are more subtle than those occurring in the phospholipase-treated permeabilized cells (discussed above) and, therefore, are more difficult to detect via conventional MS analysis.
Mast Cell Degranulation: Lipid Arrays
Comparison of the degranulation in RBL-2H3 and B6A4C1 cells shows both cell types initiate exocytosis when cells are challenged with Ca2+ ionophore (Figure 5B⇑). When we analyze these cell responses in terms of lipid profiles by MS and lipid arrays, we see two general types of response patterns. The first type of response is related to the stimulus used (i.e., antigen or ionophore), and the second is dependent on cell type (i.e., RBL-2H3 vs B6A4C1). As shown in the positive lipid array in Figure 6A⇓, some species of PC change in response to ionophore, whereas little or no responses are evoked in response to antigen. The second type of response, illustrated in Figure 6B⇓, shows reproducible changes in PI species as a result of ionophore as well as antigen stimulation in the RBL-2H3 cells. In contrast, there is little or no detection of changes in the B6A4C1 cells in response to antigen. This paradigm thus provides an opportunity to identify those phospholipids that are changing systematically with degranulation and to directly address the roles of specific phospholipid species and clusters of species in antigen-mediated degranulation. Recent studies suggest that the asymmetrical distribution of phospholipids within the plasma membrane plays a role in mast cell exocytosis (49). Data from our MS lipid arrays show alterations in membrane phospholipids that otherwise would typically not be detected because of the very small changes that characterize the physiological response. The first striking observation from the lipid arrays in Figure 6⇓ is the variety of phospholipids changing in RBL-2H3 and B6A4C1 cells treated with A23187, although more lipids change in RBL-2H3 than in B6A4C1 cells; especially noteworthy is the decrease in PI species, perhaps associated with PLC activation (hydrolysis and subsequent biosynthesis of PIP2) (Figure 6B⇓). In our mass spectral analysis, changes in multiple PI species are apparent at the earliest time points, in agreement with a report by Takenawa et al. (50) describing a rapid increase in PI turnover due to degranulation. The increase in LPC species in the degranulating cells can be attributed to the activation of PLA2 and associated with PC hydrolysis (Figure 6A⇓). Decrease in PC species (especially those containing long fatty acyl chains) is likely a result of PLD activation; increases in PA species are similarly consistent with previously observed patterns when degranulation was induced by bacterial phospholipases in a broken mast cell assay (37).
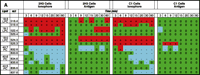

Lipidomics multiplexed analysis. Changes in individual molecular species of membrane phospholipids are shown from 2x105 cell equivalents of lipid extract from RBL-2H3 and mutant RBL mastocytoma cells (B6A4C1) for comparison. Phospholipids were extracted using the Bligh/Dyer extraction procedure under acidic conditions (46). The chloroform phase of the extraction, containing most of the phospholipids, was carefully removed and the solvent was evaporated. The resulting lipid film was immediately dissolved in CH3OH:CHCl3 (9:1), containing 1% NH4OH to facilitate protonation, and analyzed by mass spectrometry. Mass spectra were acquired on a Bruker Esquire-LC 00146 ion trap mass spectrometer (Bruker Daltonics, Billerica, MA) equipped with an ESI interface. All samples were sprayed in positive and negative mode, resulting in a variety of molecular ion signals in the range of m/z 500–1000. Excerpts from the generated positive (A) and negative (B) mode lipid arrays are illustrated. Phospholipids are presented with the class abbreviation preceded by xx:y, where xx is the total carbon atoms in the fatty acid chains and y is the number of double bonds. (See text for details.)
As shown in Figure 5A⇑, mutant B6A4C1 cells fail to degranulate in response to antigen treatment; the comparative lack of changes in membrane lipid composition of B6A4C1 cells treated with antigen seen in the lipid arrays (Figure 6⇑) is consistent with the degranulation data. There is also a different pattern of lipid changes between treatments (ionophore vs antigen) of RBL-2H3 cells, suggesting that perhaps the exocytosis triggered by these two stimulants occurs via slightly different mechanisms. These results illustrate the feasibility of assembling a lipids parts list for a cellular function of interest. Additional experimental perturbations and detailed analysis of specific lipid species are required for the assignment of particular species or subsets of species to a specific function, but the measurement of changes in phospholipid production during receptor-induced degranulation of mast cells is instructive as to the lipidomics approach.
Polyphosphoinositides
Lipidomics permits the detection and identification of important membrane signaling molecules that were previously difficult to fully characterize by other methods. This includes assessment of changes in cellular levels of polyphosphoinositides. Traditionally, PI-monophosphate (PIP) and PI-bisphosphate (PIP2) species have been analyzed as a deacylated “pool” of [32P]-labeled inositides by HPLC. This procedure can result in estimates of total PIP and PIP2, but chemical identification of the species within the PIP/PIP2 pools (i.e., species distinguishable from one another by their fatty acyl chains) in the context of other changes in lipid species has been challenging. However, using ESI-MS/MS, we have identified approximately twenty polyphosphoinositides and determined acyl content for several of these species. As shown in Figure 7⇓, agonist (zymosan) challenge of RAW 264.7 cells leads to elevated levels of a wide variety of PIP and PIP2 species compared to basal levels. Interestingly, a recent report also utilized ESI-MS to provide a detailed characterization of PIP lipid species extracted from rat brain (52).
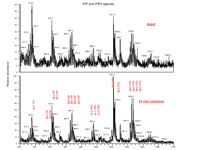
Polyphosphoinositides. Stimulation of RAW 264.7 cells with zymosan led to elevated PI-monophosphate (PIP) and PI-bisphosphate (PIP2) levels (top panel) compared to basal (lower panel). 3 × 106 cells were challenged with 50 μg/ml zymosan (a protein-carbohydrate complex derived from yeast cell wall) for 15 minutes followed by aspiration and pelleting. Inositides were then extracted using a modified Bligh/Dyer extraction procedure (51). Mass spectral analysis was performed on a Finnigan TSQ Quantum triple quadrupole mass spectrometer (ThermoFinnigan, San Jose, CA) equipped with a Harvard Apparatus syringe pump and an electrospray source. Samples were analyzed at an infusion rate of 10 μl/min in negative mode over the range of m/z 400 to 1100.
Conclusion
A full understanding of biological models of cell signaling requires detailed knowledge of membrane composition. The ability to simultaneously assess the metabolic dynamics of hundreds of phospholipid species opens a new perspective on the cellular lipome. The tight control and regulation of membrane lipid composition is manifested in subtle changes that can be probed by diverse agonists. The construction of lipid arrays to compare changes between experimental conditions provides a valuable tool for evaluating relationships among multiple lipid classes and species in conjunction with other cell component functions.
Mapping comprehensive lipid changes in time has a plethora of applications in cell biology and signal transduction. Data of this type will prove useful in hierarchical clustering schemes as a method for differentiating receptor-mediated cellular events involving lipid second messengers as well as for studying membrane compositional remodeling. In addition, these arrays form a “fingerprint” that can be used to identify cellular/ligand combinations and as such may prove very useful in diagnostic profiling.
As elegantly articulated by McLafferty and Turecek (53), “The mass spectrum shows the mass of the molecule of interest and the masses of the pieces from it.” This power to resolve individual molecules, unambiguously identify them, and detect minute changes in biomolecules in a systematic manner allows the investigator to establish relationships between seemingly distant (and often convoluted) signaling and metabolic events in cells. As substrate-product relationships are established by improvements in lipid array technology, we will be able to discriminate how distinct pools of lipid substrates contribute to distinct temporal and spatial signals that allow coordinated responses to very complex extracellular signaling. These network signaling maps will change the way we approach new pharmacological questions and help discern the mysterious processes that occur beneath the opaque world of the cellular lipid bilayer.
Acknowledgments
This work was supported by grants to H.A.B. from the National Institutes of Health (GM58516) and the Alliance for Cell Signaling (NIGMS62114). The authors acknowledge Dr. Jared S. Cohen for contributions to the mast cell degranulation work. The degranulation-deficient B6A4C1 cells were a generous gift from Dr. David Holowka at Cornell University and Dr. Reuben P. Siraganian at the National Institute of Dental Research, NIH. We are especially grateful to Ms. Michelle Armstrong for expert assistance in mass spectrometry as well as for artistic contributions to this review, and Ms. Johnna Goodman for superb editorial and administrative support.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

H. Alex Brown, PhD (back left), is the Ingram Associate Professor of Cancer Research in the Department of Pharmacology at Vanderbilt University Medical Center. Jeffrey S. Forrester PhD (back middle), is a Research Instructor trained in mathematics and computer programming. Stephen B. Milne, PhD (back right), is a Research Assistant Professor trained in analytical chemistry. Pavlina T. Ivanova, PhD (front left), is a Research Fellow with a background in synthetic organic chemistry. Michelle D. Armstrong, BA, (front right), is the Laboratory Manager, assists in the mass spectrometry analysis, and contributed to figures for this review.

