Purine Receptors: GPCR Structure and Agonist Design
Abstract
An integrated approach to the study of drug-receptor interactions has been applied to adenosine receptors (ARs) and P2Y nucleotide receptors. This approach includes probing the receptor structure through site-directed mutagenesis and molecular modeling, in concert with altering the structure of the agonist ligands. Goals of this structural approach are to generate a testable hypothesis for location of the binding site and subsequently to enable the rational design of new agonists and antagonists. In this manner, receptor subtype selectivity has been increased, and agonists have been converted into partial agonists and antagonists. An approach to receptor engineering (neoceptors) has been explored, in which synthetic small molecule agonists (neoligands) are specifically tailored to activate only receptors in which the putative binding sites have been modified. This orthogonal approach to receptor activation, intended for eventual gene therapy, has been demonstrated for A3 and A2A ARs.
Introduction
Research into the structure and function of G protein–coupled receptors (GPCR) represents a major gateway to the future of rational therapeutics. GPCR structure may be probed indirectly, by evaluating the relative abilities of a series of ligand structures to bind or activate the receptor [i.e., structure-activity relation-ships (SAR)], or directly, by altering GPCR structure through mutagenesis (1–3). Both of these approaches are greatly aided by molecular modeling, which to date continues to rely on the only GPCR structure that has been solved, namely, that of rhodopsin (4). Eventually, the elaboration of data that localize and characterize the ligand binding sites of GPCRs will be used in the rational design of new agonists and antagonists (Figure 1⇓) as well as in the engineering of GPCR structures that will engender novel pharmacological properties.
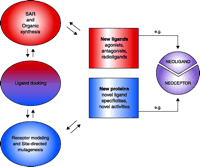
Integration of drug design and receptor modeling. Experimental data [e.g. structure-function relationships (SAR)] reveal interactions between multiple sets of ligands and receptors (i.e., both wild-type and mutant proteins); rational refinement of receptor (by mutagenesis) and ligand design (by organic synthesis) can then be suggested by ligand docking. The two stages—ligand design (red) and receptor design (blue)—are combined in an iterative manner to culminate in new ligands (e.g., agonists, antagonists, and new pharmacological probes) that will selectively bind, fail to bind, or modulate wild-type or mutant receptor proteins. A special integration of the two stages can culminate in “neoligands” that selectively bind to engineered “neoceptors.” It should be noted that GPCR modeling currently depends on the high-resolution structure of bovine rhodopsin (resting state) as template (4).
We have integrated these approaches to study drug–receptor interactions in the cases of adenosine receptors (ARs) (5) and P2Y nucleotide receptors (6), two classes of seven-transmembrane helical receptors that couple through G proteins. In addition to serving as model systems for elucidating GPCR functionalities in general, ARs and P2Y receptors are important in their own right as targets for drug development.
Extracellular adenosine acts as a local modulator of cell function via four AR subtypes (A1 , A2A , A2B , and A3 ) that are involved in numerous physiological and pathophysiological processes (5). For example, adenosine attenuates the damaging effects of ischemia in the heart and brain; acting through the AR subptype A2A , it suppresses inflammation (7) and causes vasodilation and inhibits platelet aggregation, thus increasing the amount of oxygen available to organs under stress. Adenosine agonists selective for the A3 AR are of interest as cerebroprotective (8), cardioprotective (9, 10), and anticancer (11) agents.
Adenosine derivatives have in recent years been used extensively as lead structures for the synthesis of compounds that function as selective AR agonists (12,13). AR antagonists are most commonly xanthines or other non-nucleoside heterocycles; however, recent strategies have explored the structural modification of nucleosides to obtain compounds that act as AR antagonists or partial agonists, especially at the A3 subtype (14–19). Previously, the conversion of GPCR agonists into antagonists, although carried out quite successfully, beginning with the pioneering work of Ganellin and coworkers on histamine derivatives (20), had been performed on an empirical basis. Current strategies, however, benefit from detailed structural knowledge of drug–receptor interactions at atomic resolution.
Extracellular purine nucleotides (e.g., ATP and ADP) [and pyrimidine nucleotides (e.g., UTP and UDP)] also function as signaling molecules (e.g., as neurotransmitters) that modulate the function of diverse mammalian cell types and tissues under both normal and pathophysiological conditions. The metabotropic (i.e., G-protein coupled) receptors of such nucleotides, denoted as P2Y receptors, occur as eight distinct subtypes; nucleotide- gated ion channels, denoted as P2X receptors, have also been characterized (6, 21). The distribution of P2Y receptors is broad, and they elicit considerable therapeutic interest as modulators of the gastrointestinal, cardiovascular, and immune systems (22, 23) as well as in the treatment of cystic fibrosis and other pulmonary diseases (24). Antagonists of P2Y1 and P2Y12 receptors are of therapeutic interest in modulating platelet function (25, 26).
The design of agonists and antagonists of GPCRs is limited by the current state of homology modeling (3, 12), and in particular by lack of a suitable structure of activated rhodopsin that could serve as a template for agonist docking at GPCRs of interest. In addition, the pharmacological implications of receptor dimerization and the presumed stoichiometry of GPC–G protein interactions (i.e., one receptor dimer per α subunit) are largely unknown (27). Indeed, the agonist-like state is (at least) a three-member complex: GPCR plus G protein plus agonist. Furse and Lybrand (28), for example, have cautioned that de novo modeling may be more reliable than rhodopsin-based modeling for docking of agonists to the β-adrenergic receptor. Nevertheless, the current methodology has provided useful insights into ligand–receptor interactions (29). Thus, a rhodopsin-based approach having the ability to relax the conformation of amino acid side chains during the docking has provided pharmacologically consistent docking models for agonists at the purine receptors (12). In this approach, the wealth of muta-genesis data and a wide range of agonist/antagonist structures are essential to guide the docking of agonists by selectively applying constraints within the binding pocket. Such docking is intended to represent a transient binding mode rather than a fully activated conformation of the complex.
This review describes recent advances both in the design of ligands, including those locked in specific conformations (30, 31), derived from adenine nucleoside and nucleotides, and in the rational mutagenesis of receptors (32, 33), which has resulted in proteins that display unique pharmacological properties (e.g., constitutive activity) (34). Finally, we discuss the concept of neoceptors (35), engineered receptor variants that are selectively activated (i.e., relative to the wild-type receptor) by rationally synthesized small molecules (“neoligands”).
A3 Adenosine Receptor
Of the four AR subtypes, the most recently discovered A3 AR appears to have the most restricted localization in the body, which is advantageous for pharmaceutical development. Structurally diverse antagonists have been discovered for this particular AR, although agonists are currently the focus of a surge of interest for therapeutic treatment. For reasons of expediency, the pharmacological characterization of new A3 AR agonists was initially studied using binding assays (13), which provide rapid SAR results but do not directly assess receptor efficacy with respect to each new ligand. More recently, it has in fact been established that receptor efficacy, along with ligand affinity, is highly dependent on minor changes in nucleoside structure (36), and the intrinsic efficacy of adenosine derivatives in activation of the A3 AR is more variable than at other subtypes (15). Such variability among AR subtypes raises the caveat that agonist derivatives that are used as pharmacological tools must not be assumed to act consistently across a range of receptor subtypes. For example, N6 -cyclopen-tyladenosine (CPA) and 2-chloro-N6 -cyclopentyladenosine (CCPA) have been established as a pair of potent agonists at the A1 receptor subtype. A comparison of their functional effects, on phosphoinositide turnover and forskolin-stimulated cyclic AMP production in Chinese hamster ovary (CHO) cells expressing the human (h) A3 AR, however, shows that whereas CPA remains an agonist at the A3 AR, CCPA is a full antagonist (36). Thus, those nucleoside–protein interactions that mediate the binding per se of ligand can be distinct from interactions that directly support the activation of the A3 AR (Figure 2⇓).
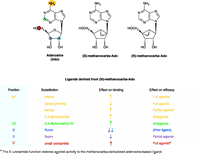
Differential effects of ligand derivatized on binding affinity and functional efficacy. The substitutions of adenosine as tabulated below the structures are offered as general examples of effects that can be observed from ligand derivitization. (Yellow indicates the N6 -position; green, C2-position; blue, 2′-position; purple, 3′-position; red, 5′-position.) The ribose moiety of the adenosine (Ado) ligand becomes effectively fixed in either the (N) or (S) conformation upon replacement with the methanocarba ring system as shown. (For a conformational representation of the (N)-fixed ring system, see the chemical structure shown in Figure 4.) Because the A3 AR prefers the (N)-conformer >100-fold relative to the (S)-conformer, the (N)-methanocarba derivative of adenosine has been used to synthesize novel A3 AR ligands of high affinity (some of which are shown in Figure 3). The (N)-methanocarba modification alone improves ligand binding to the A3 AR and reduces functional efficacy of the receptor, resulting in a partial agonist. [See (30, 35, 39)].
Structure–efficacy relationships have been studied by modifying various regions of nucleoside structure [i.e., the N6 , (29, 32), C2, (27, 31, 33), and ribose regions (14, 16)]. Specific groups placed at the N6 -position and on the ribose moiety may reduce or completely abolish the ability of ligands to activate this receptor without compromising high affinity. Similar to previous studies of the N6 -position, various 2-substituents can decrease the intrinsic efficacy of A3 AR activation. Moreover, certain N6 -substitutions (such as cyclopentyl and 3-iodobenzyl) combined with the 2-chloro modification can obliterate efficacy at both human and rat A3 ARs (36).
Modification of the ribose moiety of nucleotides and nucleo-sides provides new insights into the structural and conformational requirements for ligand functionality at purine receptors (36). The ribose ring can adopt a range of conformations, as described by a “pseudorotational cycle” (37). One approach to “freezing” the ribose-like moiety of receptor ligands in receptor-preferred conformations is to synthesize a cyclopentyl derivative that is sterically constrained through the inclusion of a second, cyclopropyl ring (38); in this way, the “methanocarba” ring system indicated in Figure 2⇑ has been employed to restrict the ribose moiety to either a Northern (N) or Southern (S) envelope conformation. Figure 3⇓ schematically illustrates the difference is between (N) and (S)-methanocarba isomers. As with certain P2Y receptors (39), ARs demonstrate a preference for ribose analogs constrained in the (N) conformation (40).
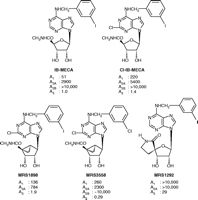
Various flexible and ring-constrained derivatives of adenosine that serve as A3AR probes. N6 -(3-iodobenzyl)-5′-N-methylcarbamoyladenosine (IB-MECA)] and its 2-chloro analogue are widely used agonist probes. The full agonist MRS1898 is a conformationally-constrained (N)-methanocarba equivalent of Cl-IB-MECA. In the antagonist MRS1292, the 5′-amide moiety, which otherwise when freely flexible guaran-tees full agonism at the A3 AR, is also conformationally constrained. Ki values (nM) in binding to human A1, A2A, and A3 ARs and functional potency at the hA2B AR are shown (30, 31, 35). See text for details.
Whereas the A3 subtype binds preferentially to the (N) con-former (Figure 3⇑), the “frozen” ribose moiety actually hinders receptor activation (resulting in partial agonism), an inhibitory effect that is overcome by addition of a uronamide group at the 5′ position [e.g., 5′-N-ethylcarbamoyladenosine (NECA) and N6 -(3-iodobenzyl)- 5′-N-methylcarbamoyladenosine (IB-MECA)] (Figure 3⇑) (36). In other words, a conformational change in the ribose moiety of the already bound nucleoside, especially in relation to the richly H-bonding 5′-uronamide group, is likely required in order to activate the A3 AR.
MRS1898, the (N)-methanocarba equivalent of 2-chloro-IB-MECA, manifests a Ki value of 1.9 nM in binding to the hA3 AR expressed in CHO cells and functions as a full agonist (36). Binding assays confirm the selectivity of MRS1898 for the hA3 AR versus hA1 and hA2 subtypes, although Cl-IB-MECA is even more selective. New (N)-methanocarba analogs in this series, such as MRS3558, are over 1000-fold more selective for the A3 AR, relative to the A1, A2A and A2B subtypes; the presence of the 5′ N-methyluronamido moiety, moreover, endows members of the series with full agonism (31).
Features of the A3 AR implicated in ligand binding and in receptor activation have been explored using site-directed mutagenesis, homology modeling, and ligand docking (32, 35). Moro et al. (41) have recently elaborated a unified pharmacophore model that predicts ligand characteristics, in terms of charge and steric bulk, that will effect antagonist docking into the putative A3 AR binding site. Having identified important recognition elements and conformational requirements of nucleoside ligands, we are using molecular modeling to further explore the steric and electronic requirements for binding from the perspective of the ARs (35, 12). The docking of several agonist ligands into the putative binding site of the A3 AR model can be accomplished in a manner consistent with pharmacological observations (17, 18, 36). The hydrophilic ribose moiety, for example, becomes surrounded, through implementation of docking algorithms, by polar amino acid side chains in the vicinity of transmembrane domains (TMs) 3 and 7, while the less polar adenine moiety interacts with the hydrophobic regions of TMs 5 and 6. Although a template for the activated state of the receptor is unavailable, the docking of adenosine agonists into the resting state model of the A3 AR provides useful information about the atomistic complementarity of the ligand with specific amino acid residues in the putative binding site.
The mode of docking of nucleosides in this putative binding site is corroborated by mutagenesis results, as validated for specific interactions using neoceptors (32, 35). A neoceptor (see below) displays a selective enhancement of affinity of a structurally modified nucleoside, when a complementary functional group is added to the receptor protein. This gain of function is indicative of spatially relevant and energetically favorable interactions between the ligand and its receptor. Whereas mutagenesis of A1 and A2A ARs has been performed (12, 42, 43), only recently have single amino acids been probed for functionality in the A3 AR (32). A number of residues in TM3, TM6, and the second extracellular loop (EL) have been individually replaced. Some of these residues [e.g., His95 (3.37), Trp 243 (6.48), Ser 247 (6.52), Asn 250 (6.55), and His272 (7.43)] had been implicated, by analogy to previous molecular modeling studies of other ARs, in ligand recognition. (The GPCR numbering system, in parentheses, indicates the TM and, following the decimal point, a residue number relative to a conserved amino acid within the Group A GPCR family, thus allowing comparison of location among diverse GPCR sequences (2).] The N250A mutant receptor fails to form a stable complex with either agonist ([125 I]I-AB-MECA) or antagonist ([3 H]PSB-11); the Asn 250 side chain putatively accepts an H-bond from the amino group of adenine. The H95A substitution significantly reduces affinity of both agonists and antagonists. In contrast, the K152A (in EL2), W243A, and W243F mutations do not significantly affect agonist binding but decrease antagonist affinity approximately 3- to 38-fold, suggesting that these residues define a general binding region for high-affinity A3 AR antagonists. Thus, putative h A3 AR binding regions for agonist and non-nucleoside antagonist appear to be overlapping but not identical, which is indicated through ligand docking in AR homology models (12, 32).
Furthermore, it has been possible to separate the structural bases for binding and activation processes by monitoring the activation of phospholipase C by wild-type and mutant receptors. The A3 agonist Cl-IB-MECA stimulates phosphoinositide turnover in cells expressing the wild-type receptor, but failed, even at concentrations as high as 100 μM, to evoke a response in cells expressing W243A or W243F mutant receptors. Nevertheless, the binding affinity of Cl-IB-MECA (Ki = 2 – 3 nM) is unchanged by replacement of Trp 243 . Thus, Trp 243 is required for A3 AR activation (and for binding of antagonist) but not for binding of agonist. The conserved aromatic function that is supplied by Trp243 also plays a role in activation of other GPCRs, including the A2A and P2Y1 receptors (32, 12).
Hypotheses for conformational changes involved in activation of ARs have been proposed. Using a rhodopsin-based model for ligand docking, we have proposed that a movement of the conserved Trp 243 may be involved in A3 AR activation (36). In particular, agonist binding appears to require rotation of the indole side chain of Trp243 , whereas antagonist binding manifests no such requirement; in the resting state or the A3 AR, Trp 243 is important for constraining the intramolecular network of TMs through both hydrophobic and H-bonding interactions. The activational rotation of the Trp 243 side chain is counterclockwise as viewed from the extracellular side, consistent with a proposed rotation of TM6 upon activation of other GPCRs (44). Mechanistically, this rear-rangement upon agonist binding may destabilize the ground-state structure by disrupting the intramolecular TM network. It has been speculated that the conserved Pro 245 might act as a flexible hinge for the agonist-induced unbending of TM6, leading to the subsequent rearrangement of TM7, IL3 and helix 8. These regions are important for signal transduction through the G-protein as proposed in the activation of rhodopsin (1–3, 45).
The observation of decreased A3 AR efficacy of adenosine derivatives upon specific structural changes (Figure 2⇑) has made possible the design of nucleoside-based antagonists. One advantage of such antagonists over non-purine A3 AR antagonists is that they overcome the limitation of selectivity to only certain species, characteristic of the non-purine antagonists (41), which tend to be orders of magnitude more potent at the hA3 AR in comparison to rodent species. Thus, it has been possible to design nucleoside-based antagonists (36), which in many cases are selective for both human and rat A3 AR. Such A3 AR antagonists (Figure 2⇑) include those adenosine derivatives that bear steric constraints of the ribose-like moiety (36), N6 -substitutions (32), or a combination of substituted N6 -benzyl groups along with various small substituents at the adenine 2-position (17). A radically altered analog in which the properly functionalized adenine moiety was shifted from the 1′-position to the 4′-carbon proved to be an A3 AR selective antagonist (16).
MRS1292 (Figure 3⇑), a ring-constrained form of IB-MECA, is a full antagonist of the A3 AR. This nucleoside analog incorpo-rates the 5′-uronamide into a spirolactam ring, attached through the ribose 4′-carbon (36). This rigid nucleoside analog maintains high selectivity (>300-fold) in binding to the A3 AR; however, it is incapable of inducing the activation process. This result further emphasizes that flexibility in the 5′-region of ribose is a prerequisite for A3 AR activation, perhaps in concert with rotation of TM6 (36). A3 AR antagonists, such as MRS1292, have demonstrated utility in models of glaucoma, in which activation of an A3 AR on the non-pigmented retinal epithelial cells promotes the accumulation of fluid in the aqueous humor (46).
P2Y1 Receptor
Similar to the study of the ARs, structurally important features of P2Y receptors are defined on the basis of sequence analysis, homology modeling, and ligand docking (Figure 4A⇓) (33). In order to ascertain which residues of the P2Y1 receptor are involved in ligand recognition, individual residues of both TMs (e.g., TM3, -5, -6, and -7), as well as ELs (e.g., EL2 and -3), have been replaced through missense mutations. A Lys residue in TM6 and two Arg residues in TM3 and TM7 (Figure 4B⇓), all located at the exofacial side of the helical bundle, putatively coordinate the phosphate moieties of nucleotide agonists and antagonists (33). Identification of these residues has allowed us to effectively model docking of ligands at the P2Y1 receptor.
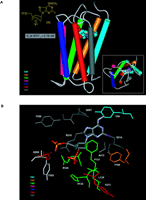
Docking of the high-affinity antagonist MRS2500 at the human P2Y1receptor. A. The basis for the conformational constraint is the methanocarba ring system, in which cyclopentane and cyclopropane rings are fused. The placement of this ring fusion at the 4′ carbon assures that a North (N) conformation is maintained. The large enhancement of affinity produced by this specific conformation within the P2Y receptor family is limited to the P2Y1 receptor (with the possible exception of P2Y14 , which is not yet characterized) (27). “P” represents a monophosphate group. The inset shows the docking as viewed from the extra-cellular side. B. A more detailed image indicates three basic residues, Arg 128 (3.29), Lys 280 (6.55), and Arg 310 (7.39), that putatively coordinate the phosphate moieties (49). The importance of these three amino acid residues in recognition of nucleotides is supported by mutagenesis studies (32). The residues Arg128 and Gln 307 occur roughly one helical turn below the exofacial side of TMs 3 and 7, respectively. [See also (32, 49).]
The principle of conversion of native agonist into antagonist has also been applied to the P2Y1 receptor. Unlike the case of the A3 AR in which this transformation is dependent on pendant functionality of the adenine and rigidification of the ribose, with the P2Y1 receptor it is dependent on repositioning of phosphate moieties. In particular, we have extended an initial finding by Boyer et al. (47), namely, that separation of the 5′-diphosphate moiety into separate monophosphate groups (i.e., at 5′ and either 2′ or 3′ positions of the ribose moiety) reduces agonist efficacy. Molecular modeling has enabled optimization of antagonist design not only through perturbation of receptor-phosphate interactions, but also through functional substitution of the adenine component of P2Y1 receptor ligand (48); in regard to the latter, we found that only limited steric bulk is tolerated at the N6 position, consistent with subsequent findings from receptor modeling (33).
A focus on sugar puckering has led to the first selective P2Y1 receptor agonist (i.e., MRS 2365, the (N)-methanocarba analogue of 2-methylthio-ADP) and antagonist (i.e., MRS 2500, the (N)-methanocarba analogue of 2-iodo- N6 -methyl-2′-deoxy-adenosine-3′,5′-bisphosphate) with subnanomolar affinities (30, 49). The docking of MRS 2500 is shown at the human P2Y1 receptor in Figure 4⇑. The use of carbocyclic analogs of the ribose moiety, as in MRS2500, is in accord with molecular modeling analyses that the ribose ring oxygen does not participate in direct interaction with the receptor (50); the possible role of the sugar as a “spacer group” between phosphate and adenine moieties and a comparison of cyclic versus acyclic nucleotide antagonists were also undertaken (48). The position of the two phosphate groups in the receptor-docked conformation appears to be key to high affinity of antagonists (Figure 4B⇑) (51).
Biological characterization of the novel P2Y1 ligands has been carried out in platelets and other cell systems, and antagonism of the P2Y1 receptor has been introduced as a new concept in antiplatelet therapy (25, 26, 52, 53). The affinity of various nucleotide analogs as P2Y1 antagonists correlates well with the inhibition of platelet aggregation. MRS2365, which is a highly selective agonist for the P2Y1 receptor, induces shape change in human platelets without causing aggregation, thus confirming that the P2Y1 subtype is associated with the shape change and that activation of both P2Y1 and P2Y12 receptors is required for full aggregation induced by the native, nonselective agonist ADP (49). The use of MRS2365 as a selective pharmacological probe to establish the specificity of P2Y1 receptors is based on the (N)-methanocarba ring system (39, 49); other ring-systems used to constrain nucleotide analogues were not as effective for delineating P2Y1 selectivity or for enhancing ligand affinities (51).
Receptor Engineering: Neoceptors
The clinical use of adenosine agonists as cytoprotective agents (5, 49) has been impeded by the broad tissue distribution of ARs, which greatly complicates the side effect profiles of exogenously administered adenosine derivatives. For example, experimental A1 AR agonists that had been intended to target brain neurons in the treatment of cerebral ischemia (54) also cause bradycardia by targeting the same receptor subtype expressed in the A-V node of the heart; tissue-specific prodrug schemes to circumvent such side effects have not yet proven fruitful. We have therefore explored the possibility of engineering, through site-directed mutagenesis, AR molecules that are characterized by novel agonist/antagonist specificities. The successful protein engineering of such “neoceptors,” in concert with the implementation of proper ligand (i.e., “neoligand”) design, could be a first step toward “neo-gene therapy” protocols. “Proper” neoligands in this context will naturally be typified by reduced potency at wild-type receptors. The general concept of engineered GPCRs (i.e., neoceptors) has been demonstrated for the hA3 AR, as well as the hA2A AR (Figure 5⇓) (35).
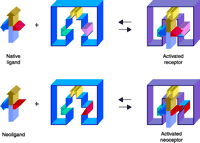
The neoceptor concept. Three hypothetical recognition elements on the receptor are schematized along with their complementary functional groups on the agonist ligand (34, 12, 53). Note that red and pink functional groups on the ligand and receptor are modified in a concerted fashion.
Manipulation of the sequence of the A3 AR through site-directed mutagenesis has led to pharmacologically unique constructs (i.e., neoceptors and constitutively active mutant receptors) that may later prove to have potential for cardioprotection (10) through organ-targeted gene transfer. Molecular modeling suggests that His 272 , which resides in TM7 and likely interacts with the 3′-hydroxyl group of the ribose moiety of adenosine, as a site for A3 AR mutation. The H272E receptor mutant proves in fact to be selectively activated by adenosine derivatives that were modified in a chemically complementary fashion; 3′-amino-3′-deoxy derivative of adenosine binds to this mutant in a selective fashion (35). Amino groups placed at other positions on the nucleosides do not enhance binding. In general, amino groups placed on the ribose moiety pharmacophore tend to reduce affinity of agonists at the wild-type ARs, whereas certain amine substitutions selectively enhance binding affinity at certain mutant ARs.
This approach has also been demonstrated at the anti-inflammatory hA2A receptor (12, 55). Neoceptors were constructed to bear anionic residues in TM3 and TM7, so as to interact complementarily with amine functions placed on nucleoside analogs; adenosine derivatives were accordingly functionalized at the 5′-, 2-, and N6 -positions. Intriguingly, the T88D mutant protein produced according to this strategy manifests enhanced binding of the chain-length–optimized 5′-(2-aminoethyl)uronamide but not the 5′-amino-5′-deoxy analog, suggesting that the amino group of the former, but not the latter, can attain proper geometry for interaction with the novel Asp88 carboxylic group (Figure 6⇓). A standard, potent adenosine agonist NECA is inactive at the T88D mutant receptor. Amino groups placed near the nucleoside 2- or N6 -position only slightly affect the binding to the mutant receptors. Thus, we have identified and modeled pairs of neoligand and A2A AR–derived neoceptor that are pharmacologically orthogonal with respect to the native species.
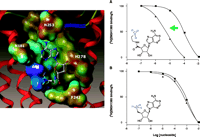
Selective enhancement of the amine-bearing neoligand MRS 3366. The human A2A AR is strategically mutated [T88D (3.36)] to provide an acidic side chain for electrostatic complementarity (A) to an amine-functionalized adenosine derivative MRS 3366 (12). Placement of the amine group closer to the ribose moiety in 5′-amino-5′-deoxyadenosine (B) fails to result in an enhancement. The receptors were expressed in COS-7 cells. In the radioligand binding competition curves shown, the squares represent wild-type, and the triangles T88D mutant receptors. [3H]ZM241385 is a potent antagonist radioligand of the A2A AR. [The color range of the electrostatic potential Connolly surface calculated using the MOLCAD module of SYBYL® is displayed from red (most positive) to blue (most negative). The nucleoside is represented by ball-and-stick model with atom-type-color. The hA2A T88D neoceptor is displayed as a tube model using the MOLCAD ribbon surface. A portion of EL2 is visable at the top of the figure.]
Conclusions
The design of novel ligands for purine and pyrimidine receptors, in some cases based on a rational, structural approach, has result-ed in selective receptor probes that promise to be useful in pharmacological studies. These probes can display enhanced potency and/or selectivity, and in some cases stability, over previously used ligands. The geometry of receptor docking of antagonists has been correlated with binding affinity, and the dependence of activation on regional conformational flexibility of both agonist and the receptor is evident.
Advances in the general methods of molecular modeling and in the resolution of the template structures of rhodopsin have brought this integrated perspective to a stage of practicality as a medicinal chemical approach to studying GPCRs and purine/ pyrimidine receptors in particular. Molecular models of the A3 AR and P2Y1 receptor have provided insights into the determinants of ligand recognition at putative binding sites. Identification of microscopic complementarity between specific receptor residues and the docked high-affinity ligands allows us to define energetically stabilizing elements in ligand recognition, which has aided in the design process. The design of mutant A3 ARs having unique pharmacological features, including neoceptors that are activated selectively by tailored synthetic neoligands, has been accomplished and may bring a new dimension to the future of gene therapy.
The ribose ring has been a recent focus in medicinal chemical efforts to alter the potency and efficacy of nucleoside derivatives. In general, the (N)-conformation of the ribose-like moiety is favored over the (S)-conformation for interacting with the P2Y1 or A3 adenosine receptors. This applies to affinity observed for both agonists and antagonists. The new, potent A3 AR agonist MRS3558, partially constrained in a receptor-preferred conformation, takes advantage of this effect. At A3 ARs, the efficacy of adenosine derivatives is reduced by certain modifications, such as 2-chloro or N6 -iodobenzyl substituents and ribose constraints, leading to selective antagonists such as MRS1292. Highly specific ligands of subnanomolar affinity at the hP2Y1 receptor have also been obtained by focusing on the ribose conformation.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Zhan-Guo Gao, PhD, is a Staff Scientist in the Molecular Recognition Section, Laboratory of Bioorganic Chemistry, NIDDK, NIH. After his medical studies, he became interested in the mechanisms of action of GPCRs, especially adenosine receptors.

Stefano Costanzi, PhD, completed his doctoral studies in medicinal chemistry at the University of Camerino, Italy, with Prof. Gloria Cristalli. Currently, he is a member of Dr. Jacobson’s research group at the NIH. His main interest is the study of the structure, function, and ligand interactions of GPCRs by means of computational techniques.

Soo-Kyung Kim, PhD, is a Visiting Fellow in the Molecular Recognition Section at the NIDDK/NIH. She is a computational chemist and has been studying the homology modeling of adenosine receptors, the docking of subtype-selective agonists and antagonists, and the design of neoceptors and neoligands.

Kenneth A. Jacobson, PhD, is Chief of the Molecular Recognition Section of the Laboratory of Bioorganic Chemistry, NIDDK, NIH. He is a medicinal chemist, with interests in the structure and pharmacology of GPCRs, in particular receptors for adenosine and purine and pyrimidine nucleotides. Dr. Jacobson currently serves as Chair of the Medicinal Chemistry Division of the American Chemical Society. Address correspondence to KAJ. E-mail kajacobs{at}helix.nih.gov; fax 301-480-8422.

