Mast Cells: The Missing Source of Cardiac Renin?
The renin-angiotensin system (RAS) plays a critical role in cardiovascular function. Traditionally, the RAS is viewed as a circulating endocrine system (Figure 1⇓) . In such a system, renin from the kidney is released into the plasma and cleaves angiotensinogen into angiotensin I (Ang I). Ang I is subsequently converted to the biologically active peptide angiotensin II (Ang II) by angiotensin-converting enzyme (ACE). Ang II is the major effector peptide of the RAS and is involved in cardiac hypertrophy and fibrosis. These cardiac effects of Ang II are partly the result of increasing blood pressure. In addition, Ang II may modulate cardiac structure and function directly by activating angiotensin receptors expressed in cardiac tissue (1).
In recent years, the concept was established that a few organ systems, including the heart, possessed a local RAS. Central to this idea is that Ang II can be produced locally and can function as a paracrine or autocrine factor. In fact, the local production of Ang II in the heart has been shown experimentally: the measurement of cardiac angiotensin peptide production, through the use of radiolabeled peptide precursors, suggested that most of the Ang I and Ang II found in the heart is produced locally, rather than derived from the circulation (2). This would require a local source of the other essential RAS components. Today, it is generally accepted that both angiotensinogen and ACE can be synthesized in the heart (1); however, the cardiac expression of renin remains controversial.
The expression of cardiac renin mRNA has been reported as either undetectable or expressed in low amounts (3, 4). Others have reported that the heart exclusively expresses a truncated isoform of renin (of unknown function) that is sequestered in the mitochondria rather than being secreted (5). It is apparent, however, that de novo renin expression does not account for most cardiac Ang I production––cardiac production of Ang I and Ang II was largely abolished in bilaterally nephrectomized animals (6–8). Furthermore, forty-eight hours after bilateral nephrectomy, only ~1% of normal renin activity remains in the heart, and in isolated heart preparations, Ang I and Ang II production cannot be detected except if external renin is infused (9). Thus, it is generally believed that the uptake of renin from plasma is the major source of cardiac renin, although the exact pathway for renin uptake is still under debate (10). The uptake may occur either by renin diffusion into cardiac interstitial spaces, or by binding of renin with various renin-binding proteins.
In a recent article published in The Proceedings of the National Academy of Science, Silver et al. detected renin protein in a subgroup of cardiac interstitial cells, and subsequently identified these granulated cells as mast cells (11). Renin expression was determined by immunostaining of rat heart sections using a polyclonal renin-specific antibody, the specificity of which was meticulously verified. First, Silver showed that the histochemical signals could be eliminated if renin pre-absorbed antibody was used. Next, they demonstrated by immunoblotting that the antibody did not cross-react with cathepsin D, a lysosomal enzyme that shares 60% amino-acid sequence similarity with renin and is also expressed in mast cells. Double labeling of heart tissue with both renin-specific and cathepsin D–specific antibodies showed that the two antibodies stained the same cells, but in different sub cellular compartments. Therefore, the authors established that the antibody directed against renin specifically stained renin. By morphologic criteria, the renin-positive cells resembled mast cells. To verify the identity of the cells, tissue sections were stained by toluidine-blue, staining the basophilic particles that are characteristics of mast cells. To further confirm that the renin positive cells were indeed mast cells, the authors used specific staining by histamine-specific antibody and found that histamine staining overlapped renin staining. Thus, the authors established that cardiac tissue contained a mast-cell population, and that renin was present in these cells. Renin staining was not found in any other cells of the heart.
To show that mast cells directly synthesized renin (rather than absorbing renin from the circulation), the authors measured renin mRNA expression in the human mast cell line, HMC-1. By using RT-PCR, they showed that HMC-1 cells did express human renin mRNA. Renin protein expression in HMC-1 cells was verified by immunoblotting. Furthermore, the renin activity was measured by testing the efficiency of cell degranulated proteins to convert angiotensinogen into Ang I. Because HMC-1 cells also express cathepsin D, which has renin-like activity, the authors verified renin-specific activity by conducting the experiment with or without a specific renin inhibitor. Under such conditions, they showed that 70% of total Ang I formation could be attributed to renin activity. Hence, the authors demonstrated that mast cells express biologically active renin.
Finally, the authors studied the spatial distribution of mast cells in rat heart tissue and concluded that mast cells were juxtaposed to nerve endings. With such a tissue distribution pattern, Silver et al. suggest that degranulation of mast cells during ischemia–reperfusion may play a pivotal role in activating local Ang II formation, thus stimulating Ang II receptors on nerve endings and increasing release of norepinephrine. The authors speculate that mast cells may be a potential therapeutic target for controlling a hyperactive RAS and excessive catecholamine release in heart disease such as congestive heart failure. Mast cells may also participate in the local RAS function in other organs.
The report by Silver et al. is the first to show renin expression in mast cells. Given the important role of the RAS in cardiac disease and the current debate concerning renin expression in the heart, the discovery of renin in cardiac mast cells is definitely interesting. It is also surprising because the detection of renin in cardiac cells was rarely reported by other groups, even when similar immunostaining techniques were used (12, 13). For example, Iwai et al., showed that a subpopulation of macrophages and monocytes in the heart bound renin-specific antibodies (13). In the study by Iwai et al., renin-specific antibody stained some cells that infiltrated the fibrotic tissues encompassing necrotic myocardium; these cells were identified as macrophages by their positive staining with an antibody directed against the CD11b/c antigen (a commonly used macrophage marker). Similar to Silver et al., Iwai et al. also performed the experiment in vitro and detected renin mRNA expression in peritoneal macrophages; however, they could not detect renin-expressing cells in normal hearts, nor could they find renin expression in cells other than macrophages or monocytes.
One reason that other research groups did not find renin expression in cardiac mast cells is the low level of renin expression by these cells. The renin protein level in HMC-1 cells was evaluated by immunoblotting and was much lower than the level of renin protein expression in the kidney. Low amounts of renin in mast cells were also demonstrated by the renin activity assay reported in the article by Silver et al. After degranulation, HMC-1 releases proteolytically cleaved Ang I (from angiotensinogen), of which approximately 70% was renin-specific. This actually demonstrated a rather low amount of renin considering that the other renin-like enzyme in mast cells, cathepsin D, is 105 times slower than renin in its angiotensinogen-cleaving efficiency. With such a low level of expression, together with the relative small mast-cell population in heart, mast cells might not be a major source of cardiac renin activity. This supposition supports the idea that renin uptake from circulation is the major source of cardiac renin. In addition, renin activity in mast cells appears to be located in the granules, suggesting that it may only be activated after mast cell degranulation. Thus, it is possible that mast cell renin may not play a significant role in a local cardiac RAS under normal physiologic conditions.
This is not to say that cardiac mast-cell renin has no significance in cardiac function and disease. As the authors suggest, cardiac mast cells are in close spatial relation with synapses, thus they may modulate local RAS activity in their immediate environment and influence the activities of cardiac nerves. Previously, mast cells were also reported to be associated with cardiac vasculature (14). Moreover, mast cells may further accumulate in the heart under disease conditions such as ischemic–reperfusion injury, cardiac infarction, and heart failure (15, 16). Therefore, mast cell renin may play a significant role at or near nerves and blood vessels, especially during the course of cardiac disease. Evidence suggests that mast cells may mediate cardiac injury. Specifically, studies have been carried out in mast cell–deficient animals to dissect the specific role of cardiac mast cells (16, 17). For example, a c-kit mutation in rat, which disrupts the gene encoding the stem-cell factor receptor, prevented the formation of mature mast cells from their precursors. These rats developed diastolic dysfunction with age in the absence of imposed cardiac injuries (16, 17). In a similar mutant mouse model, mast cell deficiency prevented the progression from cardiac hypertrophy to heart failure in a pressure overload model (17). In this experiment, treatment with tranilast, a mast cell–stabilizing agent that inhibits degranulation, mimicked the effects of mast cell deficiency, suggesting that mast cell degranulation may contribute to the progression of heart failure. It is uncertain, however, whether any of these effects are mediated by renin release, because vast amounts of other bioactive substances––including cytokines, chemokines, growth factors, metalloproteinases, and other proteases––are released during mast cell degranulation.
It is worth noting that mast cells are also a well-known source for cardiac chymase, an enzyme that is capable of converting Ang I into Ang II, thus mimicking ACE activity. This, too, suggests that mast cells may participate in the regulation of local Ang II formation. The physiological significance of chymase in cardiac Ang II production, however, is highly controversial. In vitro studies have attributed up to 90% of cardiac Ang II production to chymase in human heart (18). On the other hand, this result cannot be repeated by most in vivo studies (19). Most likely, chymase may not exert its ACE-like activity under physiologic conditions because of the presence of natural protease inhibitors in the interstitial fluid (20). In the past few years, dozens of publications have linked chymase activity to the progression of heart failure and arrhythmia (21); however, these effects may also be a result of chymase-dependent activities other than Ang I conversion. Alternatively, similar to mast-cell renin, chymase may be a more significant player in a local cardiac RAS under disease conditions.
Silver et al. also hypothesized that mast cell renin may have functional significance outside of heart tissue. Mast cells are widely distributed in many organs, especially areas important for host defenses, such as in the skin and throughout the respiratory system. Consequently, they may theoretically contribute to a local RAS effect in these organs. In recent years, Ang II has been increasingly recognized as an inflammatory factor (22). Thus, instead of acting as a vasoconstrictor, Ang II may participate in the inflammatory processes of mast cells in these tissues; however, caution needs to be taken before generalizing renin expression from cardiac mast cells to all mast cells, because mast cell populations are highly heterogeneous, as evidenced by different mast cell properties in individual tissues, as well as across species (23, 24).
In evaluating the studies of Silver et al., the central observation is renin expression by cardiac mast cells and the suggestion that these cells may provide the renin essential for the local generation of the angiotensin II. Although this is the most notable finding of the manuscript, ultimately it also is the weakest in the sense that the paper lacks substantial proof as to the ultimate physiologic significance of the observation. There is no investigation of the role of mast cells during any form of cardiac injury. Nor is there any investigation of in vivo physiologic consequences of conditions leading to mast cell degranulation. Unfortunately, the in vivo consequences of this work will not be easy to establish. Scientists are still debating the relative physiologic importance of the local generation of angiotensin II several years after the idea of a tissue based RAS was first popularized. Even more surprising, there is no clear consensus as to the pathophysiologic role of cardiac angiotensin II in heart disease, let alone any discussion as to whether it is generated locally or due to systemic formation. Add to this, the difficulty of exploring renin release by mast cells as distinct from the significantly larger amounts of renin that infiltrate the heart via the circulation, and one begins to appreciate the difficulty that Silver and colleagues will have in truly establishing the physiologic role of renin expression in mast cells. Nonetheless, their article is a valuable contribution. It provides excellent histologic confirmation of renin expression by cardiac mast cells. Even more important, it emphasizes the presence of cardiac mast cells and poses the general question of how the vasoactive substances secreted by theses cells may influence both normal and abnormal cardiac function. If one may end with an appropriate turn of phrase, Silver et al. have “nailed their colors to the mast” in indicating their belief of an important role of mast cells in cardiac physiology. Perhaps they are right; however, substantial additional work with animal models will be necessary to establish––or even to come close to establishing––the true in vivo relevance of renin production by cardiac mast cells.
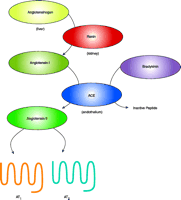
The renin-angiotensin pathway (RAS). This figure emphasizes only part of the complexity of the RAS. Angiotensin-converting enzyme (ACE) both creates a vasoconstrictor (angiotensin II) and eliminates a vasodilator (bradykinin). Cathepsin D has weak in vitro renin-like activity that may be insignificant under physiologic conditions. Chymase has ACE-like activity, but the in vivo role of chymase is still under debate. AT1 (Angiotensin II Type 1) and AT2 are the seven-transmembrane-spanning receptors for the biologically active peptide angiotensin II (1).
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Kenneth E. Bernstein, MD, is the Distinguished Service Professor in the Department of Pathology, Emory University. Dr. Bernstein’s laboratory is focused on studies of the renin-angiotensin system (RAS) and its effect on cardiovascular and renal physiology. His laboratory was one of the first to isolate and clone angiotensin-converting enzyme (ACE) and the angiotensin II Type 1 (AT1) receptor. They also discovered that the AT1 receptor activates the Jak–Stat signaling pathway. His group now studies the in vivo function of the RAS using transgenic mice that expressing ACE only in selected tissues. E-mail: kbernst{at}emory.edu; fax 404-727-8540.

Hong D. Xiao, MD, PhD, is a Research Instructor in the Department of Pathology, Emory University. She studies the role of cardiac angiotensin II production in normal cardiac physiology and in heart disease. Her research interest is to define the effects of angiotensin II in the heart that are independent of blood pressure control.

