Functional Selectivity, Ligand-Directed Trafficking, Conformation-Specific Agonism: What’s In A Name?
In classical models of drug action, the primary event is binding of a ligand (L) to its receptor (R). The binding of an agonistic ligand (Box 1) to a membrane receptor leads to an intracellular effect, E, such that L + R ↔ LR → E. The concentration dependence of the effect hinges on the binding affinity of L for R. The magnitude of the effect depends on the intrinsic efficacy of L.
Quantitative pharmacology terms.
Full agonist: A ligand that binds to a receptor and leads to a maximum biological response in the system under study.
Partial agonist: An agonist that, in a given tissue under specified conditions, does not elicit as large an effect as a full agonist.
Antagonist: A ligand that binds to a receptor, does not produce a biological response, and blocks the actions of agonists.
Inverse agonist: A ligand that binds to a receptor and reduces the constitutive activity of the receptor, thereby producing an effect opposite to that of a classical agonist.
Affinity: The ability of a ligand to bind to a receptor. Expressed in terms of an equilibrium dissociation constant, Kd, in molar units with a lower value indicating higher affinity. Ki refers to the equilibrium dissociation constant for an inhibitor determined in a competitive radioligand binding study.
Potency: The concentration or amount of a drug needed to produce a defined effect. Commonly expressed as the EC50, the concentration of an agonist that produces 50% of the maximal possible effect of that agonist.
Efficacy: The ability of a drug to produce a stimulus, indicated by the maximum effect that can be produced by that drug.
There are numerous examples showing that activation of a single receptor can lead to activation of multiple intracellular signaling events, such that:
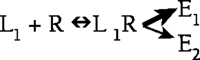
In traditional receptor theory, the relative degree of activation of E1 or E2 is the same for every agonist. For example, an agonist with half the efficacy of a reference agonist for E1 should also have half the efficacy of the reference agonist for E2. More recently, however, evidence from a variety of systems suggests that some ligands, acting at a single receptor, may preferentially activate E1 while other ligands, acting at the same receptor in the same system, may preferentially activate E2, as follows:
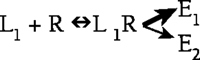
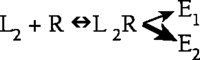
This phenomenon of ligand-dependent selectivity with respect to signaling through G protein–coupled receptors (GPCRs) was the topic of two symposia sponsored by ASPET at the 2005 Experimental Biology meeting in San Diego. The first symposium was entitled “Functional selectivity of receptor signaling: epiphenomenon or new opportunity for drug discovery?”
As described by William P. Clarke, three different processes are activated by ligands at the serotonin subtype 2C (5-HT2C) receptor, including: a phospholipase C–mediated increase in inositol phosphates, a phospholipase A2–mediated release of arachadonic acid, and receptor system desensitization. The most convincing evidence for ligand-directed selectivity of signaling in this system comes from experiments comparing the efficacy of (6)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI), 3- trifluoromethylphenyl-piperazine (TFMPP) and quipazine, all of which are serotonin agonists. When applied to Chinese Hamster Ovary cells that heterologously express the 5-HT2C receptor, DOI activates the arachadonic acid pathway with efficacy essentially equal to that of 5-HT, but elicits only 60% of the 5-HT–induced inositol phosphate response. Conversely, quipazine and TFMPP activate the inositol phosphate pathway with efficacy equal to that of 5-HT while only being able to elicit 60% of the 5-HT–induced arachadonic acid response (1).
The hypothesis arises that these differences result from distinct ligand-dependent receptor conformations. Thus, depending on the ligand, a single receptor can be placed in a conformation to either activate the inositol phosphate and arachadonic acid pathways equally, preferentially activate the inositol phosphate pathway, or preferentially activate the arachadonic acid pathway.
Some evidence suggests a role for the second intracellular loop of the receptor in mediating these differences. RNA editing of the human 5-HT2C receptor leads to amino acid changes in the putative second intracellular loop of the receptor, a portion of the receptor that is important for G protein–coupling. Ligand-dependent signaling is lost in the fully edited isoforms of the human receptor (2).
Mark von Zastrow cited three types of evidence for ligandselective functional states of GPCRs: pharmacological, biochemical, and genetic. Pharmacological evidence is exemplified by differential regulation of receptor endocytosis via clathrin-coated pits by μ- opiate receptor ligands. DAMGO [(D-Ala2, N-Me-Phe4, Gly5-ol)- enkephalin] and morphine have both been considered to be classical full agonists at the μ receptor, thus, they both activate inwardly rectifying potassium channels to the same degree. In Human Embryonic Kidney 293 cells expressing the μ-opiate receptor, however, DAMGO more effectively induces receptor endocytosis than does morphine (3). Ligand-dependent differences in opiateinduced hyperpolarization and desensitization have also been observed in rat locus ceruleus neurons (4).
Biochemical studies reveal different degrees of receptor phosphorylation in relation to differing degrees of receptor activation of μ-opiate receptors (5) and δ-opiate receptors (6). Also, recent mass spectroscopy studies of the β2-adrenergic receptor (β2-AR) show that selective ligands can determine the degree of phosphorylation of different intracellular regions of the receptor. Phosphorylation of the third cytoplasmic loop of β2-AR is increased to a similar degree by treatment with isoproterenol, epinephrine, or dopamine––three agonists at this receptor. Differences between agonists were observed in net phosphorylation of the C-terminal cytoplasmic domain of the receptor (isoproterenol ≈ epinephrine >> dopamine) (7).
Genetic evidence for functionally selective receptor conformations comes from studies of constitutively active mutants of the human complement factor 5a receptor (8). One mutant of this receptor exhibits both constitutive activation of G protein–mediated responses and constitutive endocytosis, another mutant receptor exhibits constitutive activation of G protein–mediated responses but is only endocytosed in response to ligand, and a third mutant is activated only upon exposure to ligand but is constitutively endocytosed. It is likely that such genetic variations will be important in human diseases. These variations could contribute to the etiology of a disease, to the effectiveness of drug treatment of a disease, or to individual variability in response to a drug.
David Nichols presented data showing differential signaling by agonist ligands at the 5-HT2A receptor. As with the 5-HT2C receptor, activation of this receptor results in increased inositol phosphate accumulation and arachadonic acid release. d-Lysergic acid diethylamide (d-LSD) and 1-(4-bromo-2,5-dimethoxyphenyl)-2- aminopropane (DOB) compete with binding of [125I]-2,5-dimethoxy- 4-iodoamphetamine to 5-HT2A receptors and have similar Kis, 3.5 nM (d-LSD) and 4.3 nM (DOB). A distinction between binding and signaling is illustrated by the fact that DOB increases inositol phosphates to 72% of the level produced by 5-HT and has an EC50 of 72 nM, whereas d-LSD increases inositol phosphates to only 22% of the level produced by 5-HT (d-LSD has lower efficacy) with an EC50 of 9.8 nM (d-LSD has higher potency). For the arachadonic acid responses, the relative potencies are reversed: DOB, with an efficacy 75% that of 5-HT’s, has an EC50 of 15 nM, whereas d-LSD, with an efficacy 56% that of 5-HT’s, has an EC50 of 20 nM. A dramatic difference in EC50s is seen with psilocin, which activates arachadonic acid release with an EC50 of 86 nM, but increases inositol phosphate accumulation with an EC50 of 2.3 μM (9). Data from a series of 5-HT ligands demonstrates a wide range of relative potencies for the inositol phosphate and arachadonic acid responses, suggesting that the receptor can exist in a large number of conformations, which, owing to their small differences and current limitations in our observing power, may be difficult to discern.
The 5-HT2A receptor may be involved in schizophrenia, obsessive-compulsive disorders, and cognitive disorders. It also likely mediates the effects of some hallucinogens. With respect to psychopharmacology, both hallucinogenic and nonhallucinogenic compounds were tested to see if preferential activation of phospholipase A2 over phospholipase C, or vice versa, might correlate with biological activity. Differential activation of either of these pathways by the two types of compounds was not observed, suggesting that psychotropic versus nonpsychotropic agonists, as a group, do not differ in their ability to activate selectively 5-HT2A receptor-mediated phospholipase A2 or phospholipase C signaling. Whether the generally greater potency of most ligands for activating the PLA2 pathway is relevant to the action of hallucinogenic drugs remains to be investigated.
Differential signaling through dopamine receptors was reported by Richard B. Mailman. Like the endogenous transmitter dopamine, the dopamine receptor ligands dihydrexidine (DHX) and quinpirole decrease prolactin release from the pituitary, but DHX and quinpirole do not have the same effects on striatal neurons where quinpirole is very potent and DHX is not. Such differences, observed in different cells and tissues, could be explained by a number of mechanisms, and do not necessarily arise from differences in ligand functional selectivity. The ligand dependence of this differential activation of signaling pathways was demonstrated by studies of pituitary lactotrophs, which express both short and long forms of the D2 receptor, but not other dopamine receptors. In this system, DHX acts as an agonist with efficacy equivalent to that of dopamine to increase intracellular accumulation of cAMP. In contrast to this, and unlike dopamine, DHX has no effect on G-protein–gated inwardly rectifying K+ (GIRK) channels but acts as an antagonist to block the effects of dopamine on these channels (10).
An example of the potential clinical significance of functional selectivity is provided by studies with the antipsychotic drug aripiprazole which, depending on the effect being measured, acts as a partial agonist, a full agonist, or an antagonist (11), although definitive demonstration of a relationship of this selectivity to clinical effects has not been made.
Taken together, the data presented thus far provide a considerable body of evidence showing selective ligand-induced actions through a single type of receptor, and suggest that different agonist ligands can lead to different receptor conformations. To shed some light on different functional conformations, Brian Kobilka presented studies on site-specific labeling of Cys265 of the β2-AR with a fluorescent dye. Based on structural similarities with rhodopsin, the Cys265 of β2-AR is thought to reside in the third intracellular loop of the receptor near the cytoplasmic end of the sixth transmembrane domain. This residue is situated among regions of the receptor important for G protein–coupling and for agonist binding. Fluorescence intensity of purified β2-ARs labeled at Cys265 with tetramethylrhodamine maleimide was measured as a function of time in response to different agonist ligands (12). Dopamine induces a rapid change in fluorescence as it binds to the receptor. Norepinephrine binding results in a biphasic change in fluorescence, with fast and slow components. Because the only difference between dopamine and norepinephrine is the hydroxyl group on the β carbon atom in norepinephrine, the fast phase of norepinephrine binding likely arises from the binding of the portion of the molecule common to dopamine (i.e., the catechol ring) while the slow portion reflects binding of the OH group. This hypothesis was confirmed by studies observing the binding characteristics of various catecholamine analogs and was supported by the finding that the catechol ring shows only a rapid phase of fluorescence change. The activity of these analogs was then compared in functional assays: cAMP accumulation and receptor internalization. Binding of the catechol ring alone to β2-AR does not lead to increased cAMP concentrations, but the binding of the catechol ring and the amine group (i.e., dopamine) does lead to increased cAMP. Maximal cAMP accumulation in response to dopamine is 95% of that induced by isoproterenol. Thus, dopamine is a strong partial agonist for the Gs response. In contrast, dopamine is only marginally effective at promoting receptor internalization. Norepinephrine, on the other hand, maximally activates cAMP accumulation (i.e., is a full agonist) and is significantly more effective than dopamine in stimulating receptor internalization. These findings suggest that the slow fluorescence transition may be related to coupling of the receptor to G protein–coupled receptor kinases and/or arrestins. These data demonstrate that ligand-induced receptor conformations lead to different functional responses.
It is clear from the data presented at this symposium, and from numerous other published studies, that ligand-induced functional selectivity is not an epiphenomenon and provides new opportunities for drug discovery. In addition, new mechanistic perspective for understanding drug actions are required, some of which have already been elucidated in the literature (13, 14). A variety of viewpoints on this issue were raised at another symposium with the title “Does functional selectivity/agonist trafficking make nothing sacred?”
Harel Weinstein suggested that the reason that functional selectivity doesn’t fit with the explanations of classical pharmacology has to do with our current powers of observation. We are now able to identify and test naturally occurring molecular variations (i.e., genetic mutations). We can engineer novel ligand and protein structures. We can express combinations of receptors and accessory proteins that may or may not be expressed naturally. And, we can test a large variety of structurally novel ligands. Thus, the seemingly novel phenotypes of functional selectivity simply reflect underlying mechanisms not observed in earlier studies that led to our traditional ways of thinking about drug-receptor interactions.
Bryan Roth observed that functional selectivity is not an in vitro artifact and that it is an older concept than many have realized. Roth noted that in 1985, Jim et al. (15) reported a lack of correlation between efficacy and the ability to induce contractions of venous smooth muscle of a series of α1-adernergic receptor agonists. At the time, these findings were curious but can now be explained by the concept of functional selectivity. Roth and Chuang had raised the possibility of ligand-induced selective signaling in the serotonergic system in a review article (16). A member of the audience, Michel Bouvier, pointed out that Portoghese raised the possibility with respect to opiates in the 1960s (17).
Patrick Sexton said that functional selectivity is seen as a change in the strength of receptor-effector coupling with different ligands. This can be observed as a reversal of the rank order of the potency of ligands for different effects or in the reversal of rank efficacies for multiple intracellular pathways. He also pointed out two caveats. First, one must consider the contribution of accessory proteins to phenotype. Second, he cautioned that many models and measurements are based on equilibrium states while, especially in vivo, drug action is dynamic.
A number of caveats were brought up during the symposia, one being that ligand action depends on the particular experimental system in which the observations are made. The G-protein component of the system, the cell type, accessory proteins, and the lipid environment can all affect signaling. Thus, the action of a ligand at a receptor depends upon the phenotype and physiological state of a cell. Consequently, pharmacological characterization of a ligand (i.e., full agonist, partial agonist, antagonist, partial inverse agonist, or full inverse agonist) at a single receptor subtype must be done in context of the system under study––one cannot extrapolate pharmacological characteristics from one model system to another. Sexton suggested that all of these may fall under the term functional selectivity, and that the particular basis for the selectivity must be specified as to the underlying mechanism.
Functional selectivity has implications for the drug discovery process. As discussed by K.J. Miller, a primary concern in industry is that if a single functional assay is chosen to assess activity, potentially valuable compounds may be missed if a ligand exhibits functional selectivity and if the assay is not an accurate reflection of this selectivity in terms of therapeutic effectiveness. The solution would be to screen compounds in multiple assays, with a primary emphasis on receptor binding. Functional selectivity may ultimately have a positive impact on drug development. If, through medicinal chemistry, the selectivity can be modified so that one or another action of a drug is enhanced, it may be possible to decrease adverse effects without altering, or while enhancing, therapeutic actions.
The final talk was given by M. Spedding, chairman of NC-IUPHAR, the International Union of Pharmacology Committee on Receptor Nomenclature and Classification, and led into a discussion of what nomenclature should be used to describe the phenomenon by which different ligands can preferentially activate different signals through a single receptor, once all the caveats have been removed. As indicated in Figure 1⇓, a plethora of terms are being used in this area. The precise meanings of the terms has not been established, which can lead to confusion in understanding the science. The symposia concluded with a call from Dr. Spedding for those interested in providing input on establishing clear nomenclature in this area to contact him at michael.spedding{at}fr.netgrs.com.
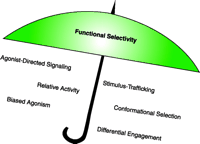
Terms that may fall under the umbrella of functional selectivity. The ability of a ligand to place a receptor in multiple conformations that lead to different intracellular signal transduction events has been described by a number of terms as indicated under the umbrella above. The variety of the terms illustrates the richness of the pharmacology that has been revealed in this area. A standardized nomenclature has yet to be defined. Figure inspired by Arthur Christopoulos.
Acknowledgments
Thanks to Bill Clarke and Bryan Roth for helpful comments on the article.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Mark A. Simmons, PhD, is a Professor of Physiology and Pharmacology at the Northeastern Ohio Universities College of Medicine. His research interests include the pharmacology of tachykinin receptors. E-mail simmons{at}neoucom.edu; fax 330- 325-5912

