SEROTONIN 5-HT2C RECEPTOR AGONISTS: POTENTIAL FOR THE TREATMENT OF OBESITY
Abstract
Obesity continues to be a burgeoning health problem worldwide. Before their removal from the market, fenfluramine and the more active enantiomer dexfenfluramine were considered to be among the most effective of weight loss agents. Much of the weight loss produced by fenfluramine was attributed to the direct activation of serotonin 5-HT2C receptors in the central nervous system via the desmethyl-metabolite of fenfluramine, norfenfluramine. Norfenfluramine, however, is non-selective, activating additional serotonin receptors, such as 5-HT2A and 5-HT2B, which likely mediated the heart valve hypertrophy seen in many patients. Development of highly selective 5-HT2C agonists may recapitulate the clinical anti-obesity properties observed with fenfluramine while avoiding the significant cardiovascular and pulmonary side effects.

Introduction
Obesity has developed into one of the most preventable causes of death in the United States and is reaching epidemic proportions in many developed nations throughout the world (1). Obesity is most often characterized by the measure of body-mass index (BMI) [i.e., weight (kg)/height (m2)]. Individuals with a BMI equal to or greater than thirty are classified as being obese, whereas those who have BMI values ranging between twenty-five and thirty are termed “overweight.” The prevalence of diabetes, hypertension, coronary artery disease, sleep apnea, cholelithiasis, and certain cancer types is highly correlated with elevations in BMI (2). Even a relatively small reduction in BMI, such as a decrease in body weight of five percent, can lead to meaningful decreases in the incidences of cardiovascular events and type II diabetes.
In the simplest terms, obesity results from an imbalance between kilocalories consumed and utilized for energy expenditure. Excess kilocalories end up being stored as fat in adipose tissue, liver, and muscle. Although controlling body weight is conceptually simple—effected by reduced caloric intake and increased energy expenditure through exercise—many people cannot achieve significant and lasting reductions in BMI. Thus, drug-based therapy may help some individuals to obtain clinically beneficial declines in body weight and a concomitant reduction in co-morbidities.
Currently available pharmacotherapies include the prescription drugs orlistat, sibutramine, and phentermine, as well as a variety of over-the-counter products (3). Most of these agents have fallen short of delivering significant weight loss and have been limited by significant side effects. The lack of highly efficacious anti-obesity agents has led to a significant drug discovery effort in the pharmaceutical industry focusing on centrally and peripherally expressed G protein–coupled receptor (GPCR) targets and enzymes involved in fat and carbohydrate metabolism (4–6). Although much of this research is in its early days, many drug targets with longer histories, such as the neuropeptide Y and melanocortin receptors, have failed to reach far enough into the clinic to establish good proof of principle (7, 8). Currently, only the CB1 antagonist rimonabant seems to be likely to enter the market in the near term (9).
Fenfluramine and dexfenfluramine achieved significant long-term efficacy in man, especially in combination with phentermine, albeit causing significant adverse drug events (10). The results suggest that a drug molecule delivering similar efficacy (via the same final mechanism), without the associated side effects, would make an attractive anti-obesity drug (11). As much of the efficacy of fenfluramine reputedly occurs through the activation of the serotonin 5-HT2C receptor, development of selective agonists for this site has been the goal of many drug discovery teams. As will be detailed below, the 5-HT2C receptor has many attributes that make it an attractive (yet difficult) target for drug development.
Fenfluramine and 5-HT2C Receptor Activation
(±) Fenfluramine (Pondimin) entered the market in the early 1970s and was followed, in the early 1990s, by the introduction of the more active enantiomer dexfenfluramine (Redux). Dexfenfluramine was the first anti-obesity drug to be approved for duration of usage in excess of three months, following a series of clinical trials making up the INDEX study (12). Redux produced only a three-percent (placebo subtracted) reduction in body weight; however, the number of patients achieving more than a ten-percent reduction in body weight was twice as great as the number doing so on placebo (11, 13) (Figure 1⇓). In addition to reducing body weight, according to evidence from some clinical trials, treatment with fenfluramine resulted in improvements to glycemic control, lipid parameters, and blood pressure (14–16).
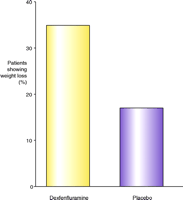
Responsiveness of patients to dexfenfluramine. The percentage of patients from each cohort (drug or placebo) showing more than ten percent loss in body weight (after twelve months) is indicated. Data are simplified from (12).
Because fenfluramine is a serotonin (5-HT) transporter substrate, it can function to elevate 5-HT levels, both by competing for uptake (transporter inhibition) and by promoting 5-HT efflux via the reversal of the transporter (releasing effect) (17). The anorectic action of fenfluramine was long presumed to be the activation of multiple receptors resulting from the increased synaptic levels of 5-HT. Indeed, several 5-HT receptor subtypes are implicated in the regulation of feeding (18). The elevated release mechanism was called into questioned, however, with the observations that neither the depletion of 5-HT stores by p-chloroamphetamine (19, 20) nor blockade of the 5-HT transporter by fluoxetine (21) inhibited the anorectic activity of fenfluramine in rats.
The desmethyl-metabolite of fenfluramine, norfenfluramine, attains concentrations about one-half that of fenfluramine in human plasma (22) and is efficacious in reducing food intake and body weight when administered to rats (19). Norfenfluramine is a potent full agonist at the 5-HT2C receptor, suggesting that much of fenfluramine’s efficacy results from 5-HT2C activation (20). Studies in rats have revealed that selective 5-HT2C antagonists can completely block the anorectic activity of dexfenfluramine (23). In addition to data pointing to 5-HT2C receptor activation as the primary basis of fenfluramine efficacy, there is a considerable amount of literature suggesting that 5-HT1B receptor activation also contributes to the acute anorectic action of the compound (5). The relevance of 5-HT1B agonism to the chronic efficacy of fenfluramine and to drug development for obesity is less clear.
5-HT2C Receptor Molecular Biology
The 5-HT2C receptor, a class A GPCR, is one of fourteen 5-HT receptor subtypes (24). Based on gene sequence, pharmacological properties, and signal transduction coupling, the receptor has been grouped with the 5-HT2A and 5-HT2B receptors into a common subfamily (25). The gene that encodes the 5-HT2C receptor is found on the X-chromosome (Xq24) and possesses four exons within the open reading frame and two in the 5′-untranslated region (26, 27). The open reading frame encodes a protein that is 460 amino acids in length. Alternative splicing may result from the use of an alternative 5′-donor site to yield a truncated receptor that can be expressed in a transfected cell line but does not bind ligand or signal through G-proteins (28). Polymerase chain reaction strategies have localized the mRNA for the splice variant in several brain regions but its function is unknown.
Homodimerization of the full-length 5-HT2C receptor has been reported in transfected cell lines, but it does not appear to be regulated by agonist binding (29). It is not yet known whether the full-length receptor can dimerize with the truncated alternative splice variant of the receptor, whether mRNA editing may affect dimer formation, or if heterodimerization occurs with other GPCRs.
The 5-HT2C receptor is the only known GPCR to undergo mRNA editing (30). Editing occurs at five positions that encode amino acids within the second intracellular loop of the receptor (31) and influences G-protein coupling efficiency (32). The level of expression of the various edited isoforms varies between brain regions. Within the human hypothalamus, the VNV isoform appears to predominate, although other isoforms are clearly present (31). Studies evaluating the chronic treatment of rats with anti-depressant drugs have demonstrated that the ratio of edited isoforms can be modulated, although not in all strains of animals (33).
Two significantly expressed polymorphisms of the 5-HT2C receptor have been identified. The C23S polymorphism is found in the N-terminal region of the receptor and occurs at an allelic rate of 0.13 (26). Studies to date have not shown any influence of this polymorphism on receptor pharmacology or on obesity (34, 35). A polymorphism in the promoter region (−759C/T) of the 5-HT2C receptor–encoding gene has recently received a great deal of attention. A number of studies have linked the polymorphism to weight gain associated with use of atypical anti-psychotics (36, 37; for review see 38), although an almost equal number of investigators have failed to see such an association (39–41). At the functional level, the −759C allele appears to result in lower receptor expression in cultured cells (42), whereas the −759T allele has been associated with greater promoter activity (43). Although Pooley et al. observed an association between the −759T/C polymorphism and weight gain, examination of mRNA from postmortem samples did not reveal any differences in 5-HT2C receptor expression (36). Clarifying the potential contribution of the −759T/C polymorphism to antipsychotic drug–associated obesity will require larger studies controlling for multiple factors.
5-HT2C Receptor Signaling
The 5-HT2C receptor was initially found to couple to the Gαq subunit of G proteins. Gαq activation leads to the stimulation of phospholipase Cβ, resulting in an elevation in inositol triphosphate (IP3) formation and subsequent intracellular calcium release (44). IP3 formation was found to occur in both brain tissue slices and recombinant cell lines expressing the 5-HT2C receptor (45). Signaling through Gαq allows for easy identification of agonist molecules using fluorescent- or aequorin-based assay systems to detect elevations in intracellular Ca2+. Kaufman et al. (46) and Clarke et al. (47) first suggested that the 5-HT2C receptor may couple to additional signal transduction pathways—cGMP and arachidonic acid, respectively—through alternative Gα subunits, and other studies have shown the 5-HT2C receptor to activate phospholipase D via Gα13 (48). Immunohistochemical studies have shown that 5-HT2C receptors can indeed interact with Gαi subunits (49, 50). The ability of the 5-HT2C receptor to couple to multiple Gα subunits raises the specter of agonist-directed trafficking and the question of whether signaling through one specific G protein might be particularly efficacious in the regulation of feeding and body weight. The complexity in receptor structure and signaling pathways poses significant challenges for 5-HT2C agonist drug discovery. Which edited isoform is most relevant in the presumed feeding centers? Is activation of one signaling pathway more efficacious in reducing feeding than another? Could a mechanism-based adverse event be avoided by preferentially signaling through a specific, optimal pathway relevant to treating obesity? A full understanding of these variables could prove essential in attempts to discover the most potent and efficacious agonist for obesity. In the absence of well-known relationships between molecular mechanisms (e.g., signaling and editing) that regulate feeding, however, an organization could become hamstrung by a multitude of unnecessary assays or—worse yet—could decide to avoid the target altogether.
5-HT2C Receptor Distribution
In all species examined, 5-HT2C receptor–encoding mRNA is found almost exclusively in the central nervous system, with the highest region of expression being the choroids plexus, where the physiological role of the receptor remains unclear. 5-HT2C mRNA is found in several brain regions of the rat, as determined by in situ hybridization, that are involved in feeding behavior, including the nucleus of the solitary tract, dorsal medial hypothalamic nucleus, paraventricular hypothalamic nucleus, and amygdala (51). All of these regions receive significant innervation from serotoninergic neurons arising from the raphe nuclei. Receptor autoradiography studies in the rat, utilizing [3H]-mesulergine, show a similar distribution pattern to that of the mRNA-labeling studies (52). In human brain slices, in situ hybridization reveals significant mRNA expression in both the hypothalamic paraventricular and ventromedial nuclei, and labeling in the amygdala is also observed (53).
Selective 5-HT2C agonists produce significant elevations in c-Fos within the arcuate and paraventricular nuclei, central amygdala, nucleus of the solitary tract, and ventral tegmental area of the rat brain (54). These same regions are also activated by fenfluramine and norfenfluramine (55). Unlike fenfluramine, however, direct-acting 5-HT2C agonists do not cause elevations in c-Fos in areas such as the cortex and striatum. Interestingly, those nuclei exhibiting 5-HT2C receptor–mediated elevations in c-Fos are also activated by compounds from other neurotransmitter systems involved in feeding, such as melanocortin-4 receptor agonists and glucagon-like peptide-1 (56, 57).
In an elegant study by Heisler and colleagues, fenfluramine was found to produce c-Fos activation of pro-opiomelanocortin (POMC) neurons within the arcuate nucleus. These POMC neurons also express 5-HT2C receptor mRNA, suggesting that the 5-HT2C receptor mediates fenfluramine’s action in this brain nucleus (58) (Figure 2⇓). In addition, the authors determined that either pharmacologic or genetic inhibition of melanocortin-4 receptors prevent the efficacy of fenfluramine, suggesting that 5-HT2C activation may regulate melanocortin signaling. Identification of 5-HT2C receptors in these cells is also significant in that it places the receptor on or in close proximity to those cells involved in glucose, insulin, and leptin signaling (59, 60).
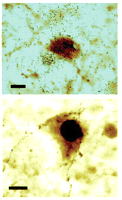
Expression of the 5-HT2Creceptor in POMC neurons active in energy regulation. After treatment with d-fenfluramine, 35S-labeled 5-HT2C receptors are apparent as clusters (upper panel) of black grains in the POMC neuronal cytoplasm (brown). The black nucleus (lower panel) reflects the immunostaining of Fos-like material following treatment with fenfluramine. Scale bar, ten microns. Excerpted with permission from (58). Copyright 2002 AAAS.
The 5-HT2C Receptor Knockout Mouse
While evidence for the involvement of the 5-HT2C receptor in feeding was emerging on the pharmacological level, the development of a transgenic mouse line lacking the receptor significantly heightened interest in agonist drug discovery. 5-HT2C knockout mice were first described by Tecott and colleagues in 1995. These mice develop late-onset obesity, weighing thirteen percent more than their littermate controls, at eleven to fifteen weeks of age, and thirty percent more after forty-two weeks (61, 62). The elevation in weight is primarily due to an increase in white adipose tissue (61). The animals are hyperphagic and exhibit metabolic hormone changes including the development of hyperleptinemia and hyperinsulinemia (61) (Figure 3⇓), and they are completely insensitive to the anorectic action of mCPP (a non-selective 5-HT2C agonist). Loss of 5-HT2C receptor expression appears to affect meal patterning, with an increase in both meal duration and frequency (62). Subsequent evaluation of these mice has shown the compound dexfenfluramine to be less effective at reducing feeding and to fail to modulate satiety sequences (63). In comparison, 5-HT1B knockout mice exhibit only a slight increase in body weight relative to littermate controls (64). These animals are insensitive to the anorectic action of fenfluramine, but recent evidence indicates this may be due to compensatory down-regulation of 5-HT2C receptor function (65).
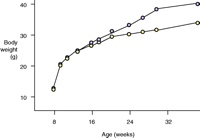
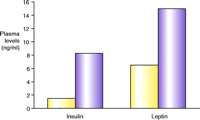
Metabolic peculiarities of mice lacking the serotonin 5-HT2Creceptor. A. The knockout mice (purple)manifest late-onset obesity, weighing thirty percent more than their wild-type litter mates (yellow)after forty-two weeks of age. B. In addition to being hyperphagic, the knockout mice exhibit metabolic hormone changes including the development of hyperleptinemia and hyperinsulinemia. Data reproduced with permission from (62). Copyright Nature Publishing Group 1998.
The Need for Selectivity in Anorectic Drug Development
A 1997 paper from Connolly et al. identified a group of patients displaying heart valve malformations that were attributed to the use of fenfluramine and dexfenfluramine (66). Subsequently, a significant number of papers appeared confirming the association of heart valve hypertrophy and the use of the fenfluramines [for review see (67)]. The relative incidence of heart valve malformations has been estimated to be anywhere between two and twenty-five percent, with longer duration of use seeming to correlate with higher incidence.
In a seminal paper by Fitzgerald and colleagues at DuPont Pharmaceuticals, it was determined that mRNAs for 5-HT2B and 5-HT2A receptors are expressed in human heart valves and that norfenfluramine is a potent full agonist for the 5-HT2B receptor and is less potent at the 5-HT2A receptor (68). The authors hypothesized that activation of 5-HT2B receptors by d-norfenfluramine (Ki = 27 nM; EC50 = 24 nM) is responsible for the valve hypertrophy, and their work has been confirmed and extended by Roth et al. (69). This group has found that interstitial cells from human heart valves express mitogenic signals following stimulation with norfenfluramine that can be blocked by a 5-HT2B antagonist (70). Other groups have also found the 5-HT2B receptor to be expressed in heart valves from human and other species, although there is some variability in which subtypes are expressed (71, 72). It should be noted that many groups have found 5-HT2A mRNA in valves from various species, and it is not possible to completely rule out this receptor as a potential contributor to the heart valve hypertrophy (71, 73).
Another significant side effect of the fenfluramines and other serotonin transporter substrates, such as Aminorex (pulled from the European market in the 1970s), is primary pulmonary hypertension (74). This often fatal condition is thought to arise from the co-stimulation of multiple 5-HT receptors on the pulmonary smooth muscle, leading to proliferation (75), and/or from the interaction with a K+ channel subtype after uptake through the 5-HT transporter (76). Whatever the exact mechanism, the need for selectivity against the serotonin receptors and transporter is highlighted.
Recent Advances in the Identification of 5-HT2C Agonists
The association of heart valve hypertrophy with 5-HT2B (and potentially 5-HT2A) agonism placed a great level of emphasis on finding potent 5-HT2C agonists that are as selective as possible. The 5-HT2B receptor is 51% homologous to the 5-HT2C receptor and is 71% identical in the transmembrane domains, whereas the 5-HT2A receptor is 49% and 80% homologous overall and within the transmembrane domains, respectively (77, 78). The fact that all three receptors utilize 5-HT as endogenous agonist suggests that the specific amino acid contact sites required for agonism will prove to be conserved. The development of highly selective 5-HT2C agonists is thus quite challenging. The compounds highlighted in this section are meant to give a flavor of the relative diversity of chemotypes being pursued and the progress in finding compounds that are truly selective (Figure 4⇓). There are undoubtedly more selective compounds in existence than the ones highlighted here but they have yet to be disclosed.
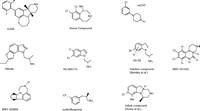
Diverse chemotypes under investigation as selective 5-HT receptor ligands. The development of highly selective 5-HT2C agonists continues to be challenging, although selectivity of certain compounds indicates that specific receptor subtypes may be targeted in anti-obesity drug development.
For many years, mCPP has been used as the prototypical 5-HT2C receptor agonist. The compound reduces feeding both acutely and chronically and produces chronic reductions in body weight (79, 80). Antagonist studies have largely demonstrated that the anorectic action of mCPP is mediated by the 5-HT2C receptor, but the compound also possesses (to varying extents) 5-HT1B, 5-HT1A, 5-HT2A, and 5-HT2B agonism (81). Thus, a number of other behavioral observations associated with the compound (both good and bad) may indeed be due to activation of multiple 5-HT receptors, especially at higher doses (82).
Ro 60-0175 is a potent non-selective 5-HT2C agonist that has been extensively profiled in animals. One study of particular interest comes from Vickers and colleagues, who administered Ro 60-0175, mCPP, and fenfluramine to Lister Hooded rats for fourteen days by mini-pump (83). Despite continuous exposure to these agonists, there was no decline in anorectic efficacy over the course of the study, highlighting the fact that the feeding and body weight reductions produced by 5-HT2C agonists do not desensitize. Although Ro 60-0175 itself is nonselective, the feeding effects of the compound have been shown to be fully inhibited by a selective 5-HT2C antagonist (84).
One of the more selective 5-HT2C agonists described in the literature is the tetracyclic indoline IL639 (Ki = 5.2 nM), with greater than 100-fold selectivity relative to other serotonin receptors (85, 4). The binding activity of this compound at the 5-HT2A (Ki = 1440 nM) and 5-HT2B (Ki = 1510 nM) receptors is comparatively minor. The heightened selectivity is presumed to be due to the addition of the biaryl headpiece to the tetracyclic core (which itself is a non-selective 5-HT2 agonist) (85). When administered orally to rats for a period of eight weeks, the compound significantly reduces food intake and body weight, demonstrating that a truly selective 5-HT2C agonist can produce efficacious weight loss (4).
WAY-163909 is a recent tetracyclic indole compound from Wyeth that shows greater than twentyfold binding selectivity for the 5-HT2C (Ki = 10 nM; EC50 = 8 nM) over the 5-HT2A (Ki = 212 nM) and 5-HT2B (Ki = 485 nM) receptors as well as over other monoamine receptors and transporters (86). The compound retains partial 5-HT2B agonist activity (EC50 = 185 nM; Emax = 40%) and lacks an appreciable 5-HT2A response. The compound has been tested in multiple rat species (86). WAY-169909, when dosed orally, reduces acute food intake in fasted Zucker rats with dietary-induced obesity. The WAY-163909–mediated reduction of feeding in Sprague-Dawley rats is blocked by a selective 5-HT2C antagonist. A ten-day chronic efficacy study with the compound in Sprague-Dawley rats shows a dose-dependent reduction in food intake and a 40% reduction in weight relative to vehicle controls at a dose of 30 mg/kg (86).
WAY-161503 is a quinoxalinone compound that is now commercially available for research purposes (87). The compound possesses exceptional potency at the 5-HT2C receptor (Ki = 3 nM; EC50 = 8 nM) and is modestly selective. WAY-163909 reduces food intake in overnight fasted Sprague-Dawley rats when given orally at a dose of 10 mg/kg.
Arena Pharmaceuticals has recently described a series of bicyclic benzazepines, of which the 8,9-dichloro compound was high-lighted (88). The compound has excellent 5-HT2C agonist potency (EC50 = 6 nM) and relatively good functional selectivity, functioning as a partial agonist of 5-HT2B (EC50 = 1800 nM) and 5-HT2A (EC50 = 220 nM) receptors. This compound and certain congeners are orally active in reducing feeding in male Sprague-Dawley rats. It is not known whether this series represents the phase-II clinical candidate from Arena that is highlighted below.
VER-3323 is an indoline derivative that exhibits modest selectivity for the 5-HT2C receptor (Ki = 24 nM; EC50 = 44 nM) over the 5-HT2A (Ki = 351 nM; EC50 = 719 nM) and 5-HT2B (Ki = 46 nM; EC50 = 11 nM) receptors, and shows excellent selectivity over other receptor subtypes (89). The compound reduces feeding in an overnight food–deprived model of food intake when given orally (MED = 30 mg/kg). Most recently, the Vernalis group has reported a series of indoles exemplified by the 7-methyl, 8-chloro compound (90), which manifests tenfold selectivity for the 5-HT2C receptor (Ki = 1.9 nM) over the 5-HT2B receptor (Ki = 19 nM) and twentyfold selectivity over the 5-HT2A receptor (Ki = 40 nM). The compound is orally effective in an overnight food–deprived feeding assay in Wistar rats.
YM-348 is a methylethylamine-substituted indazole identified by Yamanouchi Pharmaceuticals. The compound has high affinity for the 5-HT2C receptor (Ki = 0.89 nM; EC50 = 1.0 nM), for which it is fifteenfold more selective than for the 5-HT2A receptor (Ki = 13 nM; EC50 = 93 nM) but only threefold moreso than for the 5-HT2B receptor (Ki = 2.5 nM; EC50 = 3.2 nM). The compound is a full agonist at all 5-HT2 receptor subtypes (91). The compound has been extensively profiled in a number of in vivo assays, including assessments of food intake and body weight. When administered acutely, food intake is essentially eliminated in a dose-dependent manner, an effect that is blocked by a selective 5-HT2C antagonist. When given chronically for fourteen days, food intake is significantly reduced for the first week but not thereafter. Body weight is significantly reduced by day two of dosing and is subsequently maintained. Loss of white fat accounts for the majority of the weight loss. YM-348 also elevates core body temperature, raising the possibility that some of the weight loss may owe to an elevation in metabolic rate. The compound has also been found to raise mean arterial pressure, which may be indicative of its significant 5-HT2A agonism, although the classic wet dog shake seen with many 5-HT2A agonists is not observed (92).
Serotonin Receptor Modulators: Clinical Data and Perspectives
The proven efficacy of fenfluramine in the clinic supports the importance of the serotoninergic system in the regulation of appetite in man. The subsequent observation of heart valve hypertrophy and primary pulmonary hypertension, however, demonstrates that non-selective activation of the serotoninergic system in the cardiovasculature is not tolerable. The question now remaining is whether selective activation of 5-HT2C receptors will result in efficacy equivalent to that of fenfluramine in humans. The non-selective 5-HT2C agonist mCPP has been tested in acute studies in man and appears to decrease hunger ratings (93). The Arena Pharmaceuticals agonist APD-356, which is functionally more selective for the 5-HT2C receptor (EC50 = 4.8 nM) than for the 5-HT2A (EC50 = 105 nM) and 5-HT2B (EC50 = 823 nM) receptors, is currently in Phase-IIb clinical trials. The structure of this compound has not yet been disclosed, although the patent literature suggests it may be a bicyclic benzazepine. Results from Phase I and a recent Phase-IIa trial disclosed by the company (but not yet published in a peer reviewed journal) suggests that the compound reduces hunger scores and body weight following twenty-eight days of dosing and has a pharmacokinetic profile consistent with once-a-day dosing (94). Although the data are encouraging, it will take more study of this compound and other selective agonists from differing chemotypes to truly gain an appreciation of how efficacious 5-HT2C agonists will be in treating obesity in humans.
Conclusion
The 5-HT2C receptor represents a compelling target for the treatment of obesity. The localization of the receptor is consistent with a role in directly modulating pathways of food intake and body weight control. More significantly, such a role has been confirmed by the successes obtained with selective agonists thus far in pre-clinical models. The complex regulation of 5-HT2C receptor structure by mRNA editing, the ability to signal through multiple pathways, and the high homology with the 5-HT2B and 5-HT2A receptors make the identification of efficacious and selective agonists most challenging. If indeed 5-HT2C agonists can match the efficacy of fenfluramine in man, without the cardiovascular risks, they should become powerful pharmacological tools for combatting the obesity epidemic.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Keith J. Miller, PhD, is a Principal Scientist in the Obesity Department of the Pharmaceutical Research Institute of Bristol-Myers Squibb in Princeton, NJ. His current work is focused on central GPCR targets for obesity.

