Drosophila
A “Model” Model System To Study Neurodegeneration
Abstract
The fruit fly, Drosophila melanogaster, is a powerful model genetic organism that has been used since the turn of the previous century in the study of complex biological problems. In the last decade, numerous researchers have focused their attention on understanding neurodegenerative diseases by utilizing this model system. Numerous Drosophila mutants have been isolated that profoundly affect neural viability and integrity of the nervous system with age. Additionally, many transgenic strains have been developed as models of human disease conditions. We review the existing Drosophila neurodegenerative mutants and transgenic disease models, and discuss the role of the fruit fly in therapeutic development for neurodegenerative diseases.
Introduction
Neurodegenerative diseases, such as Huntington Disease, Parkinson Disease, ALS (amyotrophic lateral sclerosis), and Alzheimer Disease are devastating progressive conditions that disproportionately affect adults beyond their fifth decade of life. These and other neurological degenerative diseases are largely heritable and affect well over 100 million people worldwide, with an incidence of greater than 15% in those over sixty-five years of age. There exists no efficacious treatment for these diseases, and the prognosis is extremely poor for those diagnosed with these debilitating conditions. Cellular, pharmacological, and genetic models of these diseases have provided key information about the affected genes and pathways and will provide the basis for evaluating potential therapeutic interventions. This review will focus on the use of Drosophila as a model to study neuropathogenic mechanisms and in the development of neuroprotective compounds as therapies for these diseases.
As the world population ages, the suffering of those afflicted with progressive degenerative diseases and the associated socioeconomic costs will continue to rise. Research efforts have made important strides toward a complete understanding of these diseases; however, their progressive nature and the complexity of the nervous system are formidable challenges that hinder progress. Drosophila has emerged as an important model for understanding these diseases (1, 2). Their complex brain is capable of learning and memory and orchestrates numerous intricate behaviors. Making this possible is a nervous system composed of numerous specialized cell types utilizing all the major classes of ion channels, receptors, and neurotransmitters found in humans. Consistent with this level of neural complexity, proteomic analyses have revealed that greater than 70% of the disease-related loci in humans have a clear ortholog in Drosophila (3). The high degree of conservation revealed through proteomic analyses, the presence of a complex nervous system in an intact organism amenable to genetic manipulation, and the relatively short life-span of flies (approximately fifty days) make Drosophila ideal for studying progressive human neurological conditions.
The human brain is arguably the most complicated biological entity known. Understanding the processes that maintain the neurons that comprise the brain and how we can intervene when these processes are disrupted is a daunting task. No one scientific approach is likely to single-handedly solve all the mysteries of neurodegenerative disease; therefore, elucidating the details of neuropathogenesis associated with these diseases will surely require multidisciplinary research approaches utilizing many model systems. Although the scientific method works well for simple problems, the more complex the biological problem, the less likely it is that we are able to generate informative hypotheses. Model systems amenable to large-scale genetic screening bypass this limitation and serve the important purpose of providing novel discoveries to the field: for example, the identification of novel genes that impair neural viability with age, or the neuroprotective pathways capable of suppressing degeneration. Such approaches are capable of making discoveries that, a priori, no one could have predicted.
To date, several approaches have been utilized to isolate neurodegeneration mutants in flies (Figure 1⇓). Examples of such mutants reveal striking neurodegeneration phenotypes in the fly that enable detailed mechanistic studies (Figure 2⇓). Together, the rapid development of progressive conditions and the ability to utilize flies in powerful genetic screens support the claim that Drosophila research will continue to lead to important discoveries in this field and complement ongoing work in cellular and mammalian models.

Typical methods of neurodegeneration mutant discovery in Drosophila. Forward genetic screens (red), reverse genetic approaches (blue), and serendipity (green) constitute the main avenues of discovering this class of mutants. For each avenue, representative examples are provided. Forward genetic screens fall into three general classes based upon the method of primary screening (i.e., lifespan, behavior, or histopathology). Reverse genetic approaches are grouped by the human disease they model.

Drosophila neurodegeneration mutants. A. Na+, K+-ATPase subunit alpha mutants were identified in a forward genetic screen enriched by aberrant behavior. Aged ATPalphaDTS1 mutants (top) have severe spongiform-like neurodegeneration resulting in widespread tissue losses throughout the brain of the fly. Aged wild type control animal (A’) never exhibit spongiform neuropathology. B. Polyglutamine (Poly Q) expression causes neural degeneration in the eye of the fly. Transgenic expression of the polyglutamine expanded SCA3-Q78 results in neural degeneration seen in tangential section of the eye (top). This pathology is rescued by also expressing normal SCA-Q27 protein (B’). C. Flies lacking dADAR, the enzyme that performs A-to-I RNA editing, were found to have severe neurodegeneration (top), as compared to age-matched control animals (C’). All panels reprinted with author and publisher permission. (A and A’) Copyright 2003, Society for Neuroscience (27), (B and B’, and C and C’). Copyright 2005 and 2000, Elsevier (52) and (46), respectively.
Reverse Genetic Models OF Neurodegenerative Diseases
Numerous human diseases have been successfully modeled in Drosophila that recapitulate many key features of these diseases. These models typically involve transgenic flies expressing a human gene bearing a known dominant mutation or expressing a targeted loss-of-function mutation generated in fly orthologs of these genes. We will briefly review key features of the Drosophila Parkinson and polyglutamine (polyQ) Disease models. For a complete summary of the existing Drosophila transgenic models see Table 1⇓.
Drosophila Transgenic Models of Neurodegenerative Disease
Parkinson Disease Models
Parkinson Disease is a common neurodegenerative condition that can result from several distinct genetic mutations and specific environmental conditions. The effects of α-synuclein and parkin mutations and a pharmacological agent, rotenone, have been studied in the Drosophila model.
Important hallmarks of Parkinson Disease are the appearance of filamentous Lewy body and Lewy neurite inclusions and the selective loss of dopaminergic cells in the substantia nigra. α-Synuclein is a known component of these inclusions, and mutations in α-synuclein are known to cause familial Parkinson Disease (4). Flies overexpressing wild type or mutant (i.e., A30P or A53T) α-synuclein reveal progressive loss of dopaminergic cells in the brain (5). Transgenic α-synuclein flies, wild type or mutant, also exhibit progressive locomotor impairment, recapitulating several key features of Parkinson Disease.
Autosomal recessive juvenile-onset Parkinson Disease (AR-JP) begins in youth and is a severe form of the disease, resulting from loss-of-function mutation of parkin. The parkin protein functions as an E3-ubiquitin protein ligase, suggesting that the inability to target proteins for ubiquitin proteolytic degradation may be a direct cause of Parkinson Disease. Consistent with this hypothesis, several components of Lewy body inclusions are known targets of parkin. Flies lacking parkin function have reduced longevity, locomotor impairment, male sterility, muscle degeneration, mitochondrial impairment, and selective dopaminergic cell loss (6, 7).
Although familial forms of Parkinson Disease have led to the discovery of affected genes and important mechanistic insight into the disease, the majority of cases are sporadic, of unknown etiology, or thought to be the result of exposure to environmental toxins (8, 9). Mitochondrial complex I inhibitors, notably rotenone, are capable of causing mitochondrial dysfunction that phenocopies Parkinson Disease in vitro and in vivo (10, 11). Rotenone treatment has been used in flies to create a pharmacological model of Parkinson Disease and causes both dopaminergic cell loss and locomotor impairment (12). Additionally, Coulom and Birman show that treatment with l-Dopa and the antioxidant melatonin improve the locomotor deficit, but only melatonin is neuroprotective when coadministered with rotenone (12). One caveat with pharmacological models of disease is the difficulty in ascertaining whether the therapy is truly neuroprotective for Parkinson Disease pathogenesis or if the protection is achieved by chemically attenuating the toxin’s pathogenicity. In any event, these results are consistent with those from experiments with mammalian models of Parkinson Disease (13, 14).
PolyQ Disease Models
Many proteins contain the amino acid glutamine (Q), and some proteins naturally contain stretches of numerous glutamines, often encoded by the CAG codon. When these repeated regions are expanded to encode longer polyQ repeats, however, the result is often toxic to some neurons. This occurs in humans in the context of the huntingtin, ataxin, and androgen receptor genes and results in Huntington Disease, spinocerebellar ataxia (SCA), and Kennedy Disease, respectively. In flies, we know that expression of polyQ alone (15) or in the context of known human disease proteins such as huntingtin (16), ataxin-1 (17), ataxin-3 (18), or androgen receptor (19) all result in neurodegeneration. The dominant nature of these conditions allows one to express a polyQ-bearing transgene and to observe a phenotype without removing the function of the fly ortho-log. The eye is an ideal place to express these genes for several reasons: 1) the eye is not an essential tissue; 2) degeneration of the eye cells cause a rough appearance that can be readily observed; and 3) the eye is composed of photoreceptor cells organized into ommatidia, and quantitative measure of cell loss can be obtained by counting the number of photoreceptors remaining per ommatidium.
Studies utilizing Drosophila transgenic models of human disease have confirmed that important pathogenic features are conserved between flies and humans. The threshold for pathogenicity is similar for flies and humans: >40 Qs (20). Additionally, in flies, as in humans, the phenotypes are progressive and increased severity is associated with increased polyQ length. Another hallmark of polyQ Disease is the presence of inclusions, formed from aggregated polyQ proteins with other cellular proteins. Fly models of these diseases form inclusion bodies, the cellular components of which include polyQ proteins, chaperones, CBP [cAMP response element binding protein (CREB)-binding protein], and ubiquitin, similar to the constituent proteins found in human inclusion bodies (21, 22). Despite the different etiologies of these genetic disorders, the affected genes produce an aberration in the nervous system of the fly that is similar to the aberration in the human CNS. The common modes of pathogenesis suggest a high degree of conservation in the processes that maintain neural function with age and argue that mechanistic advances made in Drosophila will be directly relevant to the human condition.
Forward Genetic Neurodegeneration Mutants
An altogether different use of Drosophila is to utilize flies in forward genetic screens to isolate mutants that bear on a process of interest. Whereas reverse genetic approaches rely on existing genetic information, forward genetics is unbiased and is used to discover important proteins and biochemical pathways that normally function to maintain the integrity of the nervous system. To enable a forward genetic approach, one needs a reliable screening strategy to isolate the mutants of interest. The most direct method of identifying neurodegenerative mutants is to examine flies histologically for the presence of neuropathology (23). Direct histological screens are labor intensive; therefore, other researchers have reported enrichment in mutants with neurodegeneration among flies with shortened lifespan (24, 25) or with behavior abnormalities (26, 27) (Figure 1A⇑). By isolating a large collection of mutants through unbiased screens that cause neurodegeneration, scientists will discover new avenues of research by identifying disease targets or genes capable of contributing to disease pathogenesis (Box 1). Many such mutants have been identified that define pathways required for neuronal maintenance. The vast majority of these result in progressive phenotypes (conditions manifesting in adults that continue to worsen with age), like most human neurodegenerative diseases, and provide valuable insight into the pathways required for maintaining normal neural function with age. These and other methods have enabled the isolation of a large collection of neurodegenerative mutants in Drosophila (Table 2⇓).
Drosophila Neurodegeneration Mutants
The Power of Forward Genetics
Complex biological phenomena often require powerful scientific approaches to provide the initial observations that form the foundation of future inquiries. For example, forward genetic screens of mutations that disrupted embryogenesis led to the identification of numerous key biochemical pathways in Drosophila that revolutionized our understanding of animal development. The value of a large collection of mutants impinging upon “genetic control of early embryonic development” led to the 1995 Nobel prize in Medicine to Drs. Lewis, Nüsslein-Volhard, and Wieschaus. It is reasonable to believe that pathways regulating neural viability will similarly yield to a genetic approach and many such mutants have already been identified. To isolate these mutants, one needs a mutagen (e.g., chemical, radiation, or transposon) and a model system amenable to genetic manipulation. Because the whole genome is randomly mutagenized, or a large collection of transposons are screened that are randomly localized throughout the genome, and we are selecting for a phenotypic output (e.g., progressive neurodegeneration), these screens lack a bias and any gene that functions to maintain neural function with age can be identified by mutation. An important step in this process is mapping and cloning the affected gene, the speed of which has dramatically improved with the completed genome.
These mutants implicate numerous cellular functions in the normal maintenance of neuronal function with age. In some cases, the Drosophila mutant represents the first in vivo data implicating a protein’s function as essential to neural maintenance during the aging process. Although there are numerous neurodegenerative mutants, many with a novel or incomplete mechanism of pathogenesis, we will focus on those that affect neural signaling or maintenance of the plasma membrane.
Mutations Affecting Neural Signaling
Several neurodegeneration mutants have been identified that affect ion homeostasis and neural signaling. Dominant negative and loss-of-function mutations in ATPalpha cause progressive neurodegeneration and behavioral abnormalities (27). ATPalpha encodes the alpha subunit of the Na+,K+-ATPase (i.e., the sodium pump), which has an essential role in maintaining the resting membrane potential and ion homeostasis in the nervous system. Pharmacological impairment of the sodium pump can cause altered calcium homeostasis owing to reversal of the Na+-Ca+ exchanger (28, 29). Dominant negative mutations in ATPalpha also exhibit a seizure-like electrophysiological defect (27). It is possible that seizures cause the neurodegeneration in ATPalpha mutants or that seizures are caused by the same defect in ion homeostasis that leads to neurodegeneration. vacuous mutants exhibit vacuolar neurodegeneration, and a similar seizure phenotype as dominant ATPalpha mutants (26). vacuous has not been cloned but clearly maps to a distinct locus.
Heritable RNA interference has been used to generate a stable strain of flies with reduced Drosophila excitatory amino acid transporter (dEAAT1) expression (30). Decreased expression of dEAAT1 reduces the glutamate buffering capacity of flies and results in swollen mitochondria and neuropil neurodegeneration. Such a model may prove valuable in deciphering mechanisms of glutamate-mediated excitotoxic cell death in vivo.
Mutations in a trp calcium channel gene also cause severe neurodegeneration (31). Although a detailed mechanism has not been verified, calcium ion homeostasis is critically maintained in healthy cells, and dysfunction would be predicted to profoundly alter neural signaling and activate numerous calcium-dependent proteins, namely, proteases. Transgenic overexpression of the Na+-Ca+ exchanger CalX protects neurons from Trp dysfunction (32). It will be important to determine whether augmenting CalX function is neuroprotective for other ion-homeostasis mutants such as dEAAT, ATPalpha, dADAR, and vacuous.
Plasma Membrane Maintenance
Several fly mutants have been identified that demonstrate the importance of plasma membrane maintenance in neuronal viability. Adrenoleukodystrophy (ALD) is characterized by increased amounts of very long chain fatty acids (VLCFAs). In the X-linked form of the human disease there is a reduction in VLCFA acyl–CoA synthase activity leading to neural demyelination and degeneration, similar to that seen in the Drosophila bubblegum mutant (33). In flies, as in humans, dietary supplements of unsaturated fatty acids (e.g., “Lorenzo’s oil”) is an efficacious treatment for this condition.
Drosophila swiss cheese encodes a protein with sequence and functional similarity to human neuropathy target esterase (NTE), the known target of environmental toxins that cause neuropathy (34). In swiss cheese mutants, there is a lack of NTE esterase activity, and increased amounts of phosphotidylcholine are associated with widespread neurodegeneration (25, 35); similar results were found in a brain-specific NTE mouse mutant (36). Phosphatidylcholine is a major component of the metazoan plasma membrane and its concentration is elevated in Alzheimer Disease patients, suggesting these mutants may bear on a common mechanism of pathogenesis.
One caveat to forward genetic approaches is ascertaining precisely how information from these mutants will translate to mammalian systems. Some of the neurodegeneration genes identified in flies will be direct targets in human disease. Other fly genes will secondarily contribute to disease pathogenesis, and, still others may not be associated with human disease, owing to redundant gene function or early embryonic lethality in humans. For example, when Drosophila ATPalpha mutants were first discovered, the relationship to human disease was not evident. Although the Na+,K+-ATPase was hypothesized for many years to be the target of numerous neurological diseases, many of these hypotheses were disproved. This led some to suppose that this evolutionarily conserved, essential protein whose genetic locus was too highly constrained (i.e., unable to tolerate mutation) to be a human disease locus. In flies, dominant mutations and haploinsufficiency (null/+) of the Na+,K+-ATPase gene results in stress-induced locomotor impairment, reduced longevity, and neurodegeneration, suggesting that mutation of this locus may result in human disease (27). Subsequently, mutations in the human Na+,K+-ATPase gene, ATP1A2, were found to cause RDP (Rapid-onset Dystonia Parkinsonism). RDP is a devastating neuromuscular condition characterized by rapid stress-induced onset, severe locomotor impairment, reduced longevity, and Parkinson-like symptoms (37). The similarity between the phenotypes observed in haploinsufficient ATPalpha flies and the symptoms of RDP patients suggest this Drosophila model will prove valuable in elucidating the basis of this disease. This and other examples of the striking similarity in phenotypes between mutant flies and symptoms of human neurological disease suggest studies of fly mutations will help to elucidate the human condition, even when such a connection is not immediately evident.
Serendipitously Identified Neurodegenerative Mutants
Other neurodegenerative mutants have been serendipitously identified in the study of other biochemical processes. Two such examples are SOD1 (superoxide dismutase 1) and dADAR (Drosophila adenosine deaminase that acts on RNA) mutants. SOD1, also termed Cu/Zn SOD, is a broadly expressed cytosolic enzyme that catalyzes the destruction of toxic superoxides. Reactive oxygen species (ROS) cause cellular stress (i.e., damage to lipids, proteins, and DNA) and have been implicated in disease pathogenesis and aging. SOD1 mutant flies exhibit reduced longevity and neurodegeneration (38). Mutations in human SOD1 are known to cause ALS (39, 40), suggesting these flies could be used to model this disease condition.
Adenosine-to-inosine (A-to-I) RNA editing is a post transcriptional process that modifies pre-mRNA transcripts, often altering the coding potential of the processed transcripts. Although not exclusive to the nervous system, RNA editing of many ion channel or receptor transcripts have been described, and it has been hypothesized that the process contributes significantly to the protein diversity required for complex neural function in animals (41–43). Consistent with this hypothesis, the Drosophila RNA editing enzyme, dADAR, is enriched in the Drosophila nervous system (44, 45), and dADAR null mutations result in flies with severe behavioral impairment (46). Studies of dADAR-null flies also led to the discovery that RNA editing is required for the maintenance of neural integrity during the aging process in flies (46). The mechanism of neuropathogenesis in dADAR mutants is complicated because of numerous affected targets; however, loss of ion homeostasis, altered neural signaling, and ROS-dependent mechanisms have been suggested (41, 46, 47).
Drosophila AS A Model System TO Elucidate Pathogenic Mechanisms
Numerous studies have utilized various Drosophila mutants and transgenes to provide mechanistic insight into pathogenic mechanisms associated with degenerative diseases. Studies in Drosophila have implicated numerous key neuronal functions as critical to the maintenance of function during aging (Figure 3⇓). We will summarize two recent discoveries of importance that provide insight into polyQ Disease and parkin-associated Parkinson Disease pathogenesis.
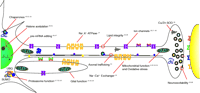
Neural dysfunction associated with Drosophila neurodegeneration mutations or transgenes. Fruit fly research has identified mutations affecting many biochemical functions essential to neuronal maintenance with age, suggesting these may be directly compromised in disease states or may secondarily contribute significantly to disease pathogenesis in humans. These mutants will enable more detailed studies of neural degeneration and senescence. Key references that bear on each process are provided. SUMO, small ubiquitin-like modifier; UBQ, ubiquitin.
PolyQ Disease Suppression
Huntington Disease, SCA, and Machado-Joseph Disease (MJD) are caused by an expansion of a polyQ-repeat region within the affected protein, leading to large aggregations within the cell (inclusions). These aggregates might arise from the misfolding of proteins and complexes formed by clustered chaperones and other proteasome components. Bonini and colleagues demonstrated that coexpression of the chaperone protein Hsp70 with the mutant MJD-expanded polyglutamine region (Q78) protein rescues the retinal degeneration caused by the expression of MJD-Q78 (18). Surprisingly, this suppression did not prevent protein inclusions within the cell but it did suppress the neurodegeneration. Another chaperone protein, dHdj1, can function in the place of Hsp70 to potently suppress MJD-Q78 toxicity (48). These chaperone proteins selectively recognize the expanded polyQ region within proteins and cause a change in the protein solubility by altering the protein’s three-dimensional structure. Bonini has presented data demonstrating that Hsp70 is also a potent suppressor of neuropathogenesis in Parkinson Disease (49). As well as mitigating polyQ disease pathology, the expression of chaperone proteins can suppress dopaminergic cellular toxicity in transgenic α-synuclein flies (49, 50). Similar findings were seen in transgenic mouse model of Parkinson Disease (51).
More recently, transgenic expression of normal human ataxin-3 suppressed the neurodegeneration caused by polyQ-containing disease proteins in Drosophila, including that caused by polyQ-expanded ataxin-3 (52). Wild-type ataxin-3 contains both a ubiquitin-binding motif and a nonpathogenic polyQ repeat and contributes to proteasome function through its ubiquitin-protease activity. Thus, wild-type ataxin-3 functions to protect against polyQ-mediated neurodegeneration, and mutant ataxin-3 is harmful, presumably owing to its expanded-polyQ protein toxicity and loss of proteasome function.
Parkin And Mitochondrial Dysfunction
Drosophila loss-of-function parkin mutants exhibit shortened lifespan, male sterility, muscle degeneration, mitochondrial impairment, and locomotor defects (6). Histopathological studies of this model of autosomal recessive juvenile-onset Parkinson Disease (AR-JP) revealed no evident pathology in the brain or loss of tyrosine hydroxylase immunostaining that would have indicated dopaminergic cell degeneration (6). Independently, another group generated parkin mutants that were remarkably similar; however, these were additionally reported to have increased sensitivity to oxidative stress (i.e., paraquat) (53). Both of these initial studies examined the dorsomedial cluster of dopaminergic neurons and found that these neurons were not affected in parkin-null animals. More detailed, careful studies of all of the major dopamine neuron clusters revealed a significant and specific loss of neurons of the protocerebral posterior lateral (PPL1) cluster in parkin mutants (7). As with the specificity of substantia nigra degeneration in humans, the specificity of PPL1 dopaminergic cell loss in the Drosophila Parkinson Disease model is not fully understood.
Recently, in flies transgenically expressing glutathione S-transferase, Pallanck and colleagues observed improvement of the locomotor impairment of flies harboring parkin hypomorphic (i.e., partial loss-of-function) alleles and rescued PPL1 dopaminergic neuron loss associated with parkin-null mutations (7). Glutathione S-transferase is thought to help neurons cope with the increase in oxidative stress. This research group also can rescue a Drosophila α-synuclein model with transgenic glutathione S-transferase expression [L. Pallanck, personal communication]. The ability to mitigate parkin- and α-synuclein-mediated degeneration suggests that the development of pharmacological methods that augment glutathione S-transferase expression may provide a viable therapy for many forms of Parkinson Disease.
Drosophila AS A Model System For Therapeutic Discovery
At the frontier of Drosophila research is the use of flies as a pharmacological system. Drosophila can be utilized as a rapid model system to test the efficacy of putative neuroprotective compounds and can even be utilized in compound screens for therapeutic discovery. A summary of important results obtained in Drosophila that provide either key mechanistic insights or represent important steps toward therapeutic development may be found in Table 3⇓.
Neuroprotective Genes and Compounds Identified Using Drosophila Model
Geldanamycin And Parkinson Disease
Mutations in α-synuclein lead to human Parkinson Disease, which is modeled in Drosophila by transgenic expression of wild type or mutant α-synuclein. Overexpression of Hsp70 can prevent the loss of dopaminergic neurons in this Parkinson model (50). Geldanamycin is thought to function by interfering with Hsp90 activity (54), which normally functions as a negative regulator of the heat shock transcription factor that mediates Hsp70 and Hsp40 expression (55). Geldanamycin prevents dopaminergic neuron loss in a Drosophila α-synuclein transgenic model of Parkinson Disease (56). Geldanamycin augments Hsp70 protein expression in vivo during cellular stress (57), which suggests that this compound may prove a viable therapy in humans.
Histone Deacetylase Inhibitors And Huntington Disease
PolyQ diseases are typically characterized by the formation of cytoplasmic and nuclear inclusions. Several lines of evidence suggest altered transcription is important to the pathogenesis of polyQ disease (58, 59). The pathogenic form of huntingtin interacts with CBP, which is known to cause transcriptional dysregulation owing to altered histone acetylation (59). Additionally, transgenic expression of CBP has been shown to suppress polyQ toxicity (58). Histone deacetlyase inhibitors such as sodium butyrate and SAHA (suberoylanilide hydroxamic acid) were evaluated for neuroprotective effects using the Drosophila huntingtin polyQ model (59). Pharmacologically inhibiting deacetylation mitigated polyQ Disease in flies. Importantly, SAHA was shown to pass the mouse blood-brain-barrier, and 2-hydroxypropyl-β-cyclodextrin–complexed SAHA (a solubilized form of the drug) improves locomotor function in the R6/2 Huntingtin mouse model (60).
Conclusions
Drosophila mutants and transgenic models have recapitulated many key features of Parkinson Disease, RDP, MJD, and Huntington Disease, and it is likely that these models will continue to be used to elucidate mechanistic details and provide the basis for evaluating drug therapies (Box 2). At the frontier of Drosophila neurodegeneration research is the utilization of flies for the development neuroprotective therapeutics. Genetic screens that exploit neurodegeneration mutants will define novel neuroprotective pathways. Mutant flies will also be used directly in pharmacological screens to identify neuroprotective compounds. Although such screens would be best suited for modest (2,000–20,000) compound libraries, the in vivo efficacy of test compounds can be efficiently evaluated using the fly model system.
Limitations Of The Fly System
Although Drosophila recapitulates many key features of human neurological diseases, with any model there are limitations. Our understanding of the fruit fly’s neuroanatomy is still developing, and it isn’t always evident which regions of the brain are performing what system level function in the fly. The fly has a blood–brain barrier (BBB) and an immune system; however, these are simple in comparison to their mammalian counterparts, suggesting that diseases associated with neuroinflammation will be difficult to model in flies and that neuroprotective compounds studied in Drosophila may need to be altered to pass a mammalian BBB. Although we point to the power of Drosophila to make novel discoveries in the field of neuroprotection and neurodegeneration, we don’t envision any model system single-handedly producing a cure for any disease. Important discoveries made in Drosophila will need to be validated in a mammalian model system, suggesting that Drosophila research should best be thought of as enabling the field by complementing work in other systems.
Acknowledgments
We thank NIH NINDS T32NS 07391-07 (AMC), the Department of Pharmacology, and the University of Pittsburgh School of Medicine for financial support; Adam Frank for assistance generating artwork; and Dr. Simon Watkins and Mark Rubin from the University of Pittsburgh Center for Biologic Imaging/Pittsburgh Center for the Environmental Basis of Human Disease (CEBHD) for access to microscopes and assistance generating images.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Alicia M. Celotto, PhD, is a PIND postdoctoral fellow and Associate Scholar in the Department of Pharmacology at the University of Pittsburgh Medical School. Dr. Celotto studies mitochondrial dysfunction, reactive oxygen stress, and bioenergetic impairment, and their role in causing neurological dysfunction. Her other passions include motorcycling, dogs, and the violin.

Michael J. Palladino, PhD, is an Assistant Professor of Pharmacology at the University of Pittsburgh Medical School. Formally trained as a molecular geneticist, his research focuses on understanding molecular details of neuropathogenesis and the development of neuroprotective therapeutics. Address correspondence to MJP. E-mail mjp44{at}pitt.edu; fax (412) 648-1945.

