Estrogen and Mitochondria: A New Paradigm for Vascular Protection?
Abstract
Mitochondrial dysfunction has been implicated as a cause of age-related disorders, and the mitochondrial theory of aging links aging, exercise, and diet. Endothelial dysfunction is a key paradigm for vascular disease and aging, and there is considerable evidence that exercise and dietary restriction protect against cardiovascular disease. Recent studies demonstrate that estrogen receptors are present in mitochondria and that estrogen promotes mitochondrial efficiency and decreases oxidative stress in the cerebral vasculature. Chronic estrogen treatment increases mitochondrial capacity for oxidative phosphorylation while decreasing production of reactive oxygen species. The effectiveness of estrogen against age-related cardiovascular disorders, including stroke, may thus arise in part from hormonal effects on mitochondrial function. Estrogen-mediated mitochondrial efficiency may also be a contributing factor to the longer lifespan of women.

Introduction
In contrast to retrospective studies (1), recently completed prospective studies of hormone replacement therapy have failed to demonstrate a vasoprotective role for estrogen (2, 3). The experimental design of these studies has thus been open to a number of concerns (4–7), chief of which is the advanced age of the women who had been studied. Given the fact that the study participants were ten or more years past menopause, studies such as the Women’s Health Initiative or the Heart and Estrogen/Progestin Replacement Study do not necessarily address vasoprotective characteristics of estrogen, because degenerative (postmenopausal) changes may not be reversible. Recent studies have highlighted a significant association between variation in the gene for estrogen receptor α (ERα) and the risk of cardiovascular disease (including stroke), underscoring a role for ERα in cardiovascular disease (8, 9). Interestingly, a recent retrospective study clearly shows that retention of the ovaries at the time of hysterectomy for benign disease increases long-term survival and decreases the incidence of cardiovascular disease (10).
Although knowledge of the non-reproductive functions of estrogen and other gonadal steroids has rapidly expanded over the last ten years, many critical questions remain to be answered, and new exciting information is still emerging. We were particularly excited, given the evidence for its protective role in cerebrovascular disease, to find that estrogen profoundly increases the efficiency of energy production and suppresses the damage of reactive oxygen species (ROS) in mitochondria (11). To appreciate the implications of the effects of estrogen on mitochondrial function, it is important to review why mitochondria are thought to play a key role in the process of aging and in the etiology and progression of a variety of age-related diseases. Although it is premature to pronounce mitochondria the winners in the “Anti-Aging Sweepstakes,” the mitochondrial theory of aging remains one of the main contenders (12).
Mitochondria: The Link between Aging, Exercise, and Diet
Metabolic syndrome, a constellation of hypertension, high plasma lipids, obesity, and glucose intolerance, has been increasing in the United States and appears to be rising in young people (13). Weight loss and increased physical activity are thought to be among the most effective measures to manage this condition. Interestingly, this is only one of a number of age-related and degenerative disorders that have been rapidly increasing in industrialized countries over the last fifty years (14). Is there a common link that could explain this rapid change in disease incidence? Whereas genetic changes in the population over this time course would be unlikely to alter health significantly, our environment has changed rapidly in the last fifty years. In particular, the abundant diet and sedentary lifestyle of humans in industrialized societies is a marked alteration that has occurred over a very short period of time.
It has been known for several decades that one of the most effective measures to prolong life is caloric restriction. Laboratory rodents maintained on a restricted caloric intake live fifty percent longer and have fewer cancers compared to animals fed ad lib (15). But why does caloric restriction prolong life, and why should there be an increase in age-related degenerative diseases in industrialized societies? A strong candidate to explain these findings occurs in hundreds to thousands of copies per cell and is directly involved in utilization of calories: Mitochondria (14). Mitochondria are believed to have evolved from bacterial symbionts and contain their own DNA (mtDNA), RNA, and protein synthesis systems. Most of the proteins that make up mitochondria are encoded by the nuclear genome, but the mtDNA itself encodes thirteen essential proteins that participate in four of the five oxidative phosphorylation complexes (see below). In addition to these thirteen subunits, the mitochondrial genome also encodes the 12S and 16S mitochondrial ribosomal RNAs and twenty-two transfer RNAs.
Mitochondria burn dietary calories in a process known as oxidative phosphorylation (OXPHOS), whereby oxygen is used to release chemical energy from food so that the body can perform work and maintain its temperature. OXPHOS is mediated by five different protein complexes (I through V) located in the mitochondrial inner membrane (Figure 1⇓). Dietary carbohydrates and fats are oxidized by the tricarboxylic acid cycle and the β-oxidation pathways, respectively, producing reduced equivalents such as NADH (reduced nicotinamide adenine nucleotide) or FADH2 (reduced flavin adenine nucleotide). The energy that is released in this controlled transfer of electrons (i.e., oxidation of metabolites) is used to pump protons from inside the mitochondrial matrix across the inner membrane into the intermembrane space through complexes I, III, and IV. The resulting electrochemical gradient serves as a source of potential energy for synthesizing ATP (i.e., phosphorylation of ADP). Importantly, a by-product of this energy production is the generation of ROS in mitochondria, and ROS can damage mitochondrial enzymes, mtDNA, and the cell in general (Figure 1⇓). More than 1% of all consumed oxygen may give rise to ROS formation (16). As a significant by-product of mitochondrial energy production, then, ROS can cause heritable damage (e.g., to mtDNA) that is perpetuated as cells divide, culminating in a progressive age-related change in function (17). ROS production increases with excess fuel supply (i.e., calories) due to an increase in OXPHOS and consequent increases in the natural by-product, ROS. ROS production also increases with decreased energy utilization (e.g., lack of exercise in skeletal muscle or lack of neuronal activity in the brain) as ATP accumulates and ATP synthase is no longer active (14, 17, 18). Oxidative stress is accompanied by inactivation of iron-sulfur centers in key mitochondrial enzymes, leading to a decrease in OXPHOS (14, 17) and a very high mutation rate of the mtDNA (19). Cells with defective mitochondria, moreover, preferentially replicate these defective organelles (14), perhaps to compensate for reduced energy production, thereby accelerating mitochondrial dysfunction with age.
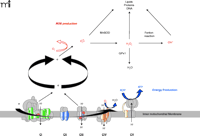
Schematic representation of mitochondrial respiratory chain. The OXPHOS system within the inner mitochondrial membrane is composed of five enzyme complexes. Reoxidation of reducing equivalents such as NADH (reduced nicotinamide adenine nucleotide) or FADH2 (reduced flavin adenine nucleotide) is coupled to the generation of an electrochemical gradient across the inner mitochondrial membrane. Mitochondrial superoxide is converted to H2O2 by Mn superoxide dismutase (MnSOD), and the resulting H2O2 is reduced to water by glutathione peroxidase-1 (GPx1). However, H2O2, in the presence of reduced transition metals (Fenton reaction), is converted to the highly reactive hydroxyl radical (OH•). Fe-S centers of enzymes in the tricarboxylic acid cycle and the electron transport chain are major targets of ROS reactivity.
In recent years, it has been shown that mitochondria play a key role in apoptosis. In response to pro-apoptotic cues, the mitochondrial permeability transition pore (mtPTP) opens, and the critical polarization of the mitochondrial inner membrane disappears as ions equilibrate between the matrix and cytosol, resulting in mitochondrial swelling (14). In addition, a number of factors are released from the mitochondrion that promote cell death. Disease states, such as cerebral ischemia, that inhibit OXPHOS and increase ROS production promote activation of the mtPTP and increase cell death by apoptosis.
Why Does Exercise or Brain Activity Slow the Progression of Age-Related Diseases?
In individuals who habitually exercise (or use their minds) ADP formation is increased because ATP breakdown increases with workload (18). Therefore, ATP synthase can continue to function and allow the mitochondrial inner membrane to remain polarized. Such freely permitted flow of electrons and concomitant ATP synthesis limit production of ROS and reduce oxidative stress. Exercise and brain activity have been shown to slow the progression of a variety of age-related diseases, including Alzheimer’s disease and type 2 diabetes (20, 21).
Characteristics of Mitochondrial Diseases Resemble Age-Related Disorders and Aging
Common characteristics of mitochondrial diseases include delayed onset and a progressive course. Furthermore, these diseases result in some of the same clinical problems found in age-related diseases and in the elderly. Specific symptoms arising from mitochondrial dysfunction include cardiovascular disease, dementias, blindness, deafness, movement disorders, muscle weakness, renal dysfunction, and endocrine disorders including diabetes (14). Mutations in the mitochondrial genome have been implicated as the cause of a variety of human diseases. The most frequently encountered mtDNA mutation is the A3243G variant in the mitochondrial tRNALeu(UUR) gene. This hot spot is known to affect mitochondrial protein synthesis and phenotypic expression (22). Interestingly, the A3243G mutation was originally found in patients with the MELAS syndrome (myopathy, encephalopathy, lactic acidosis, and stroke-like episodes), characterized by stroke-like episodes with focal cerebral hyperemia (23). Treatment of these patients with l-arginine improves the symptoms of stroke-like episodes, suggesting that pathogenesis of this condition may depend on endothelial dysfunction (24).
A plethora of evidence connects mitochondrial ROS production with aging (25). For example, the mtDNA mutation rate increases with age, and transgenic mice heterozygous for knockout of the mitochondrial (i.e., manganese-dependent) form of superoxide dismutase (MnSOD) show accelerated changes in mitochondrial function equivalent to those of much older animals (26). Whereas different isoforms of SOD localize to different cell compartments, all can metabolize superoxide, but only transgenic homozygous knockout mice defective in the gene for the mitochondrial form of MnSOD (Sod2) die within a week and show acute superoxide toxicity. This acute toxicity involves inactivation of mitochondrial enzymes containing iron-sulfur centers (e.g., aconitase and complexes I, II, and III), a phenotype that results in dilated cardiomyopathy and neurodegenerative disease causing movement disorders (27, 28). Mice heterozygous for the Sod2 gene show chronic increased superoxide production with life-long respiration inhibition and increased proton leakage across the mitochondrial inner membrane. These mice also demonstrate increased mitochondrial lipid peroxidation up to middle age, followed by a rapid mitochondrial decline that is associated with increased sensitization of the mtPTP. Later in life, a wave of apoptosis occurs in these animals that removes all of the cells with damaged mitochondria (29). These findings underscore the importance of the MnSOD enzyme exclusively to mitochondrial function. The advantages of controlling ROS damage within mitochondria have recently been supported through experiments that significantly increase the mouse life span by targeting catalase to mitochondria (25).
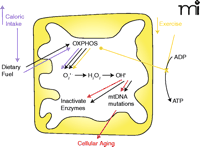
Mitochondria: Link between diet, exercise, and aging. The major role of mitochondria is to convert dietary fuel into ATP by oxidative phosphorylation (OXPHOS). As a by-product of this process, superoxide is produced within the mitochondria. Superoxide is converted by MnSOD to hydrogen peroxide and then by the Fenton reaction to a very potent ROS, hydroxyl radical. Production of ROS can cause two changes in the mitochondria: inactivation of enzymes with an iron-sulfur core and mutation of mtDNA. Accumulation of mtDNA mutations with advancing age is one mechanism of cellular aging. Increased caloric intake and decreased exercise are linked through these mitochondrial mechanisms to cellular aging. Increased caloric intake and increased dietary fuel input drives oxidative phosphorylation and increases superoxide production. This increases mtDNA mutations and increases the rate of cellular aging. Decreased exercise, by decreasing the utilization of ATP results in decreased levels of ADP, leading to stalling of the electron transport chain and diversion of electrons in the chain to produce ROS (14). Just as with increased caloric intake, increased ROS production caused by decreased exercise increases mutations of mtDNA and accelerates cellular aging.
These observations and many others not only support the mitochondrial theory of aging—the hypothesis that aging reflects cumulative oxidative damage to the mitochondria—but may also explain the effectiveness of caloric restriction in prolonging life span. With dietary restriction, transcription factors responsible for regulating mitochondrial function are activated. This leads to increased mitochondrial oxidative phosphorylative capacity and increased transcription of MnSOD and catalase (14, 18). Significantly, these types of effects have recently been demonstrated in cerebral blood vessels after estrogen treatment (10, 11).
Endothelial Dysfunction: A Key Paradigm for Vascular Disease
Endothelial dysfunction, which has been defined as the loss of endothelial vasodilator production (30), is associated with a number of disease processes, including hypertension, atherosclerosis, and diabetes (i.e., types 1 and 2). The so-called metabolic syndrome, a constellation of these disorders, has become increasingly common in the US (13, 31). Endothelial dysfunction, including loss of endothelium-dependent relaxation, has also been shown to occur with normal aging in both humans and rodents (32–34).
An important contributor to vascular dysfunction is oxidative stress (35). ROS can be generated by mitochondria (the primary source) as well as by nicotinamide adenine dinucleotide oxidase, xanthine oxidase, lipoxygenase, or (uncoupled) NO synthase. Oxidative stress has also been shown to be important in brain injury after ischemia and reperfusion. For example, in Sod2+&/minus; mice, brain infarct size is greater than in wild type after permanent focal cerebral ischemia (36, 37). Accordingly, overexpression of MnSOD results in neural protection (38). Furthermore, mitochondrial uncoupling of oxidative phosphorylation, which reduces ROS formation, protects against acute neuronal injury. Overexpression of mitochondrial uncoupling protein 2 in mice as well as chemical uncoupling of mitochondria in rats drastically reduce infarct volumes following transient focal cerebral ischemia (39–41).
Why Do Women Live Longer Than Men?
The role of estrogen in decreasing mitochondrial ROS production and, presumably, mitochondrial DNA damage, could contribute to the longer lifespan of women. Indeed, ovarian conservation at the time of hysterectomy has been shown to enhance long-term survival (10). These findings underscore concerns about over-broad interpretation of recent studies of hormone replacement therapy. Further investigation of the effects of estrogen on mitochondrial function, including elucidation of the transcription factors involved, may help to focus development on new, more selective treatments. Effects of selective estrogen receptor modulators (SERMs) illustrate the importance of the cellular milieu for the actions of nuclear receptor hormones in general (18). Thus, further understanding of the mechanisms by which estrogen affects mitochondrial function may lead to development of novel approaches to selectively affect estrogen receptor targets that protect against age-related vascular disease.
Recent studies of vascular function in Sod2+/− mice, however, have shown conflicting results. Isolated aortae from wild-type and Sod2+/− mice were comparable in terms of both contractile response to serotonin or prostaglandin F2α and in relaxation response to acetylcholine or superoxide (42). Furthermore, even after acute oxidative stress, there were no differences in vasomotor function. In contrast, measurements of flow-induced dilation in isolated mesenteric resistance arteries showed a significant decrease in the magnitude of dilation in arteries and an increase in levels of ROS from Sod2+/− mice, relative to wild-type animals (43). There are at least two ways to explain the disparity in these results. First, animals used in the first study were eight to sixteen weeks old (42), whereas animals used in the second were twenty to twenty-three weeks of age (43); deficits in mitochondrial function may have been age-related (29). Alternatively, it may be possible that changes in mitochondrial MnSOD have a greater impact in more metabolically active tissues.
Despite the pathophysiological implications of ROS, the administration of antioxidants to prevent cardiovascular disease remains controversial. Conflicting results underscore the necessity of better understanding the various sources of ROS and the mechanisms involved in their regulation. Research into this area is clearly warranted, given evidence that mitochondrial oxidative damage correlates directly with ROS levels and indirectly with lifespan (12). Certainly, a number of different factors affect the rate of aging, but increasing evidence suggests that mitochondrial oxidative stress and mitochondrial DNA damage play a major role.
Estrogen Protects Against Endothelial Dysfunction
There is considerable evidence that estrogen, either endogenous or exogenous, can prevent or delay the loss of endothelial vasodilator capacity. For example, a recent study of human arteries obtained from fat biopsies demonstrates both functional and morphological disturbances of the endothelium of arteries from post-menopausal women, effects partially reversed after incubation with 17β-estradiol (44). Some evidence, although much less than is the case for estrogen, suggests that testosterone may have the opposite effect, promoting the production of endothelial-derived vasconstrictors at the expense of vasodilators (45).
Estrogen Promotes Cerebrovascular Mitochondrial Efficiency
We have recently demonstrated that chronic treatment with estrogen has a profound impact on mitochondrial function in cerebral arteries (11, 46). These novel findings demonstrate that estrogen increases mitochondrial capacity for ATP production while decreasing generation of ROS. Because of the myriad functions of the blood–brain barrier, cerebral endothelial cells are the most metabolically active of all vascular beds (47). Thus, an effect of estrogen to preserve cerebrovascular mitochondrial function has the potential to significantly affect the occurrence and course of a number of age-related diseases, including Alzheimer’s, vascular dementia, and stroke (48).
Estrogen Receptors in Mitochondria
ERα is present in cerebrovascular mitochondria. A single protein species of 66 kDa, corresponding to ERα, is found in mitochondrial lysates of rat cerebral vessels (11). As shown in Figure 3⇓, immunofluorescence and confocal microscopy demonstrate colocalization of ERα and subunit I of complex IV (i.e., a mitochondrial marker) in the cerebral vascular smooth muscle layer. A few studies in cultured cells have also demonstrated the presence of ER in mitochondria (49–51). For example, ERα and ERβ have been localized to the mitochondria in human MCF7cells (49), whereas ERβ has been localized to mitochondria in rat primary neurons, primary cardiomyocytes, and a hippocampal cell line (50). Estrogen response elements have been found in the master regulatory region of mtDNA (i.e., the D-loop) and have also been localized within structural genes of mtDNA (52), supporting the concept that the ligand-bound ER can influence mtDNA transcription. It has been suggested that actions of estrogen on mitochondria may control cell-cycle progression of estrogen-dependent cancer cell lines (51). In MCF7 cells, exposure to estrogen increases levels of ERα and ERβ in the mitochondria and also causes a significant increase in transcript levels of the mitochondrial genes that encode cytochrome c oxidase subunits I and II (49).

Estrogen receptorα (ERα) is present in the mitochondria of cerebral blood vessels.A. A mitochondrial protein, subunit I of complex IV, and ERα are colocalized in the smooth muscle of a female rat pial artery by laser scanning confocal microscopy. Dual staining used a ERα antibody (green fluorescence) and an antibody to complex IV subunit I (red). The merged image shows co-localization (yellow). B. Enlarged image of a single smooth muscle cell (box in A) shows subunit I of complex IV (red). C. Identical cell as in B, revealing co-localization of ERα with complex IV subunit I (yellow). Only ERα (green) is present in the nucleus (asterisk). Reproduced (11) by permission of ASPET.
Estrogen Increases Capacity for Oxidative Phosphorylation
Given the presence of ERα in cerebrovascular mitochondria and the demonstration that estrogen increases mitochondrial DNA-encoded complex IV transcripts in MCF-7 cells (49), we sought to determine if estrogen treatment alters protein levels of complex IV subunits. Western blot analysis of mitochondria reveal that in vivo treatment with estrogen significantly increases both mtDNA-encoded subunit I (Figure 3A⇑) and nuclear-encoded subunit IV of complex IV (Figure 3B⇑), relative to levels in vessels from ovariectomized animals. We have also demonstrated that estrogen treatment increases mitochondrial levels of the key electron transport protein cytochrome c (11).
Two enzymes that are rate-limiting for different aspects of oxidative phosphorylation were investigated to determine whether estrogen affects mitochondrial capacity for energy production. Complex IV activity is rate-limiting for electron transport and thus for energy production (53), whereas the condensation of acetyl CoA and oxaloacetate by citrate synthase is rate-limiting in the citric acid cycle. Indeed, as shown in Figure 4⇓, both complex IV and citrate synthase activities in mitochondria from vessels of estrogen-treated ovariectomized rats are significantly greater than in ovariectomized controls. Although further studies are warranted, it is clear that chronic estrogen treatment enhances the capacity of mitochondria for oxidative phosphorylation.
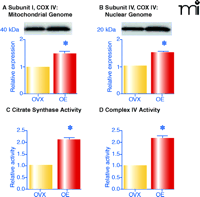
Estrogen alters oxidative phosphorylation by increasing levels and activities of key enzymes. Female ovariectomized (OVX) rats were treated in vivo with estrogen for three weeks (OE). Mitochondria were isolated from cerebral vessels and complex IV subunit I (A; encoded by mitochondrial genome) and subunit IV (B; encoded by nuclear genome) were probed by Western blot. Enzyme activities of citrate synthase (C) and complex IV (D) are also shown. Mean data are expressed as fold difference vs OVX. *p≤0.05. Adapted from (11) by permission of ASPET.
Estrogen-mediated Inhibition of ROS Production
As major producers of ROS (16, 54), mitochondria appear to function in the vasodilation of cerebral arteries by generating localized increases in intracellular Ca2+ (i.e., calcium “sparks”); a recent study demonstrates that mitochondria-derived ROS activate Ca2+ sparks and that some antihypertensive agents indeed function by activating this pathway (55). Thus, changes in smooth muscle and endothelial mitochondrial function can directly influence vascular tone. Endothelial function in particular is regulated by estrogen, and the hormone profoundly influences cerebral vascular tone (56–61). In addition to immediate effects on vascular tone, estrogen may also have longer-lasting effects involving protection against age-related changes and age-related disease. The link between mitochondrial oxidative phosphorylation, production of ROS, damage to mitochondrial DNA, and degenerative diseases associated with aging may be most relevant for understanding why mitochondrial actions of estrogen have profound implications for human aging and disease.
Given that estrogen activates mitochondrial oxidative phosphorylation, which in turn produces ROS, we became interested in the mechanisms by which the hormone promotes mitochondrial efficiency without increasing rates of ROS generation. As shown in Figure 5A⇓, we find that estrogen treatment significantly increases mitochondrial MnSOD protein in vessels from ovariectomized rats, an observation that is consistent with the literature (62). For example, estrogen treatment has been shown to increase MnSOD levels in the mouse aorta (63). Accordingly, estrogen-mediated stimulation of oxidative phosphorylation is accompanied by decreases in hydrogen peroxide in vessels (Figure 5B⇓) and superoxide levels in cultured human brain microvascular cells (HBMEC) (Figure 5C⇓). Similarly, estrogen-treated HBMEC demonstrate an increase in activity of aconitase, an enzyme with an iron-sulfur center that would be labile to oxidizing conditions (Figure 5D⇓) (64). In addition to upregulating MnSOD, estrogen also appears to increase cytochrome c levels (11), which presumably increases efficiency of electron transport without generating ROS (65, 66). Serum-induced increases in cytochrome c have in fact been shown to enhance mitochondrial respiration, even when there is no change in citrate synthase activity or complex IV subunit expression (67). By increasing mitochondrial efficiency and limiting damage from ROS, estrogen may thus have the ability to preserve cerebrovascular function with advancing age and delay the development of age-related degenerative diseases.
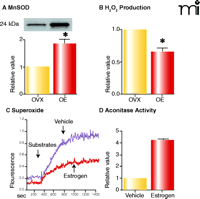
Estrogen alters ROS production in cerebral blood vessels. Western blot for MnSOD (A) and mitochondrial H2O2 production (B). Levels of MnSOD increase in cerebral vessels of female rats treated with estrogen for three weeks, whereas mitochondrial hydrogen peroxide production decreases. Mitochondrial superoxide production (C; measured using MitoSox dye) decreases in human brain microvascular endothelial cells treated with estrogen (10 nM) compared to vehicle-treated cells, whereas mitochondrial aconitase activity increases (D). Data for A and B used by permission of ASPET (11). C and D show unpublished data (Sunday, Razmara, Stirone, Krause, Duckles, and Procaccio).
How Does Estrogen Influence Mitochondrial Function?
It is clear from studies detailed above that estrogen affects mitochondrial function by modifying the expression of both nuclear- and mitochondrial-encoded proteins. In this way, estrogen has the potential to coordinate interactions between these separate genetic systems. We have found that levels of nuclear respiratory factor-1 (NRF-1), a key nuclear regulator that increases the expression of nuclear-encoded mitochondrial proteins (68), rise in cerebral blood vessels in response to in vivo estrogen treatment (11). Future studies will be important to determine the specific pathways and transcription mechanisms through which ER activation increases NRF-1 levels. Such studies may help to explore the possibility that estrogen may coordinate mitochondrial and nuclear gene expression to regulate oxidative capacity.
Implications for Development of New Therapeutic Interventions
Steroid hormone receptors, members of the nuclear receptor family, contain a conserved domain to interact with hormone-specific response elements in the genome (69, 70). In addition to this DNA binding domain, ERα activates expression of genes, using two transcriptional activation functions, AF1 and AF2. AF1 is localized to the N-terminal region of many nuclear receptors and varies significantly among different receptors. In contrast, AF2 is localized to the conserved ligand-binding domain and relies on an agonist ligand–induced protein conformation. AF1 and AF2 act either independently or synergistically in regulating gene expression, depending on the cellular and promoter context. The activation domains in ERβ are not as well understood (70).
A large number of proteins interact with the AF2 domain and enhance nuclear receptor activity (70). Some of these interact with many, if not all, nuclear receptors; however, other co-activators show tissue-specific expression (68, 71). Recent studies of the mechanisms of estrogen receptor action and the important role of coregulators have led to the concept that the ER function is dependent on cell and promoter context (72). A large number of coactivators and corepressors are thought to modulate actions of ligand-bound ERs, and a particular sequence of association and dissociation of cofactor complexes with ER-bound promoters may be a factor in determining the physiological outcome (70).
Development of selective ER modulators (SERMS), such as tamoxifen and raloxifene, has helped to elucidate the complexity of ER activation, although it is still not entirely clear how SERMS influence ERs. The tissue specificity of SERMS highlights the possibility that specific ligands might be developed to take advantage of the complexity of ER partners in order to selectively influence specific ER-mediated responses (70, 72). Better understanding of the various coregulators necessary for the actions of estrogen on mitochondrial function may make it possible in future to develop agents to selectively influence mitochondrial function.
Conclusions
The decline of mitochondrial energy production and increased oxidative stress and cell death are critical factors for age-associated impairments (14). According to the mitochondrial theory of aging, the vulnerability of mitochondria to ROS provides a link between the well-described effects of lean diets and exercise (14, 18) to protect against age-related diseases, including cardiovascular diseases. Recent findings that estrogen increases the capacity for cerebrovascular mitochondrial energy production and decreases ROS (11, 46) provide an important mechanism to explain the well-described protective effects of estrogen against age-related disorders. Endothelial dysfunction is an age-related process dependent on oxidative stress (35). Given the high metabolic rate of the cerebral endothelium as a component of the blood–brain barrier, investigation of these factors in the cerebral vasculature holds particular significance for a variety of age-related disorders, including Alzheimer’s disease and stroke.
Acknowledgments
We acknowledge NIH HL50775-12 for financial support. We are also indebted to Dr. Douglas C. Wallace and the University of California, Irvine Center for Molecular and Mitochondrial Medicine and Genetics for providing facilities, reagents, and intellectual input.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Vincent Procaccio, MD, PhD, is Assistant Professor in the Department of Pediatrics and Center for Mitochondrial and Molecular Medicine and Genetics at the University of California, Irvine. His research interests center on investigating the role of mitochondria in neurodegenerative diseases, but more recently he has also focused on the effect of estrogen on mitochondrial function.

Christopher Stirone, PhD, completed his graduate training, in 2005, in Pharmacology at the University of California, Irvine in the laboratory of Sue Duckles and Diana Krause. He is currently a Postdoctoral Scholar in the Department of Pharmacology and Center for Molecular and Mitochondrial Medicine and Genetics.

Diana N. Krause, PhD, is Professor of Pharmacology in the School of Medicine, University of California, Irvine. She trained as a neuroscientist at UCLA School of Medicine and the City of Hope. Throughout her career she has been particularly interested in the function of the cerebral circulation. For the last decade, her work has focused on the effects of gonadal steroids on cerebrovascular function.

Sue Piper Duckles, PhD, is Professor of Pharmacology and Associate Dean at the University of California, Irvine. Trained as a pharmacologist, her research focuses on the unique characteristics of the cerebral circulation and the impact of gonadal steroids on vascular function. Address correspondence to SPD. E-mail spduckle{at}uci.edu; fax 949-824-4855.

