The Fight Against Tamoxifen Resistance in Breast Cancer Therapy: A New Target in the Battle?
Breast cancer is the most common cancer diagnosed in women in the United States, with approximately 180,000 new cases per year, and some 40,000 deaths (1). Most breast cancer patients are post-menopausal women, with approximately three-fourths of all breast cancers diagnosed in women older than age fifty, and the median age of death at sixty-nine years.
Seventy per cent of diagnosed breast cancers express Estrogen Receptor alpha (ERα) and are, therefore, likely to be hormone-responsive. The most common therapy for ERα–positive cancers has employed the use of selective estrogen receptor modulators (SERMs) such as tamoxifen. SERMs compete with estradiol, the natural hormone, for binding to the ligand binding domain (LBD) of ERs. SERMs are also mixed agonists: they block estrogenic growth signals in some tissues (e.g., breast) while maintaining estrogenic responses in other tissues (e.g., bone). The other broad class of compounds in use, aromatase inhibitors (AIs), block the CYP19-encoded, aromatase enzyme that converts androgens to estrogens, thereby “starving” the ER LBD from an activating ligand. AIs are only used in post-menopausal women, where the major source of estrogen production occurs via aromatase activity in peripheral tissues, for example in breast, as the relatively vast and feedback-controlled ovarian production of estrogens has ceased.
The ER is a member of a larger group of steroid-activated transcription factors, consisting of modular domains (2). A central DNA binding domain (DBD) contains two zinc fingers, followed by a flexible hinge region and then the LBD (Figure 1⇓). Both the N-terminal domain and the LBD have transactivation functions to stimulate transcription of estrogen-regulated genes. SERMs such as tamoxifen and raloxifene induce a bound conformation in the LBD that is less likely to interact with coactivators and is more likely to associate with corepressors, thereby inhibiting ER function (3, 4). Coactivators are a family of proteins that amplify transcription by binding to the hormone-responsive activation function (AF-2) domain of nuclear receptors (5, 6). These coactivators have common nuclear receptor interaction motifs (LxxLL, NR box) and bind over Helix 12, a key structural component within AF-2, resulting in full transcription at estrogen target genes (4, 7). X-ray structures of ERα LBD (3, 4) show the NR box interaction site for coactivator binding to AF-2 is only formed with bound estradiol, not tamoxifen or raloxifene. It is also of note that tamoxifen is an antiestrogen in the breast, whereas it demonstrates relative estrogenic properties in the bone and endometrium, where more of the coactivator SRC-1 is present (8). Hence, the coactivator/corepressor profile of a specific cell-type can influence the relative ligand activity.
A large clinical trial in the 1990s tested the chemopreventive aspects of tamoxifen (9). The trial was conducted in women at elevated risk of breast cancer, either by age (older than 60 years) or for younger women with sufficient risk factors including family history, early menarche, late menopause, nulliparity, etc. The trial showed that approximately 50% of breast cancers were prevented by tamoxifen (relative to placebo) in both pre- and post-menopausal women. It had not been clear that pre-menopausal women with significant circulating estrogen would respond as well to the competitive inhibitor tamoxifen. The benefit in pre- and post-menopausal women came almost entirely in a reduction in the number of ER-positive tumors, a finding consistent with ERα as the target of tamoxifen action. Tamoxifen was also able to reduce by 50% the number of early, pre-invasive cancers [e.g., ductal carcinoma in situ (DCIS)], and it appeared to maintain bone density. Negative side-effects included increased endometrial cancers in post-menopausal women, increased blood clots, and increased cataracts. A recent clinical comparison between tamoxifen and raloxifene (STAR trial) (10) shows that raloxifene is equally effective at preventing breast cancers, with fewer side-effects, and is already approved by the FDA for clinical use to prevent osteoporosis.
As with many drugs, resistance to tamoxifen therapy for breast cancer will develop over time; also, there are still other patients whose tumors are ER-positive but do not respond to initial tamoxifen therapy. Presumably in many of these patients, ER bound with tamoxifen sometimes exhibits a partially activated conformation of the LBD that can result in estrogenic growth signals (11). Several mechanisms of tamoxifen resistance appear to include: 1) overexpressed coactivator proteins (AIB1 gene amplification leads to excessive ER activity and gene products driving growth (12); 2) under-expressed corepressors [a decrease in nuclear receptor corepressor (NCoR) correlated with tamoxifen resistance in a mouse model of human breast cancer (13)]; and 3) activated growth factor pathways that lead to phosphorylation of ER and ligand-independent growth signals. A number of reports show increased growth factor signaling in breast cells replaces the need for ER-mediated growth signals (14–16). This finding is also exhibited in later stage breast cancers with high amounts of HER2/neu, an epidermal growth factor receptor (EGFR)-related tyrosine kinase with potential to activate proliferation signals. HER2/neu is the target of successful trastumuzab (Herceptin®) antibody therapy for late-stage breast cancers that are also most often ER-negative and hormone independent (17).
Resistance to one SERM is followed, in clinical practice, with the use of another SERM or, alternatively, an AI. For example, if tamoxifen resistance appears, often a switch to fulvestrant (ICI 182,780; Faslodex®) is recommended (18), as it is a “pure” antiestrogen whose mechanism of action appears to lead to degradation of the ER (19). The mixed agonist activity and eventual resistance to tamoxifen and other SERMs probably is dependent on the continuing DNA binding potential of ER bound by those compounds. Because SERMs will continue to be used clinically for receptor-positive cancers in pre-menopausal women, one proposal addresses multiple SERM resistance by alternating low levels of apoptosis-inducing estradiol therapy, followed by fulvestrant and an AI (20).
Alternatively, a recent paper from Farrar and colleagues (21) describes a unique mechanism to alter the activity of the ER in the case of tamoxifen resistance. This research describes the use of an electrophilic compound, disulfide benzamide (DIBA), that targets preferentially the C-terminal zinc finger of the ER DNA binding domain (22), rather than the SERM targeted LBD. DIBA interaction prevents the ER from binding estrogen response elements (EREs) near estrogen regulated genes—as shown by EMSA and by transcriptional reporter studies—and thus, much of ER action in growth signaling is blocked. These authors used co-immunoprecipitation experiments to show that DIBA may restore tamoxifen sensitivity to cells that were resistant by inhibiting AIB1 coactivator binding and by increasing NCoR interaction with tamoxifen-bound ER. In a xenograft tumor model in mice, DIBA alone, and more so in combination with tamoxifen, did inhibit growth of human tamoxifen resistant tumors derived from MCF-7/LCC2 cells. These results are perhaps the most promising that DIBA may increase the antiproliferative properties of SERMs to shrink tumors in vivo, especially after resistance has developed. Does DIBA lead to increased degradation of ERα, similar to the action of fulvestrant on the LBD? If so, although the experiment was not performed, this could effectively reduce the amount of ERα in the cell that transmits tamoxifen-resistant growth signals.
It remains to be seen if DIBA will be a useful compound in the clinic. There are several papers outlining such electrophilic compounds as antiviral agents that react with a required zinc finger structure in the coat proteins of HIV, inhibiting viral infection (23). Until clinical trials are done to test DIBA in combination with tamoxifen or another SERM, it remains unclear whether other proteins or tissues will also be targeted, with potentially negative consequences. DIBA is not yet available in a form that can be taken orally.
From recent clinical trials, there appears to be no subgroup of postmenopausal hormone-responsive breast cancers for which AIs (e.g., letrozole, anastrozole, or exemestane) are not at least as effective as tamoxifen (24–28). There are fewer side-effects than with tamoxifen and there is an improved profile of disease-free survival. At least in postmenopausal women, in whom most breast cancers occur, AIs are now the recommended first-line treatment (29–31). Though AI therapy can lead to increased bone loss during its use, other alternatives, such as bisphosphonates, appear to render this manageable. Therefore, secondary agents to restore tamoxifen sensitivity, such as DIBA, most likely will remain for use in a smaller number of pre-menopausal breast cancer patients.
However, as a proof of principle, DIBA is very interesting compound because it targets a second functional domain of ER. Its targeting of the DBD not only allows a potential increase in efficacy for tamoxifen, but probably for any of the SERM class of molecules that target the LBD. As such, perhaps these compounds can be of major importance for ER-positive, hormone-responsive or -resistant breast cancer therapy in pre-menopausal women, once some of the clinical trial and safety data are determined.
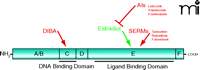
Treatments for ER+ breast cancer and chemoprevention that target specific domains of the estrogen receptor. Estradiol activates estrogenic growth signals after binding the estrogen receptor. Two major classes of compounds are used in breast cancer therapy to inhibit ER growth signals. The aromatase inhibitors (AIs) inhibit the enzyme responsible for production of estradiol from androgenic precursors. Selective estrogen receptor modulators (SERMs) compete with estradiol for binding to the ligand binding domain and alter the activator or repressor proteins that subsequently bind. Disulfide benzamide (DIBA) acts in a novel way, by interrupting the second zinc finger of the DNA binding domain, preventing receptor interaction at EREs.
Acknowledgments
The author would like to thank Drs. Victor G. Vogel, Pamela A. Hershberger, and A. Paula Monaghan for giving comments and helpful suggestions about the manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2007
References:

Mark Nichols, PhD, is an Assistant Professor in the Department of Pharmacology, University of Pittsburgh, and a member of the University of Pittsburgh Cancer Institute, Molecular Therapeutics and Drug Discovery Program. The research interests of his laboratory include study of steroid hormone receptors, primarily the estrogen receptors (ERα and ERβ), and their role in normal, as well as in cancer tissue. Better understanding of ligand activation of ER may lead to improved endocrine therapies for treating and preventing breast cancers while preserving positive aspects of estrogen signaling in other tissues. Investigation of a number of compounds that bind ERs, resulting in various biological outcomes, is ongoing to define new therapeutic targets and pathways. E-mail: mnichols{at}pitt.edu; fax 412-623-4840.

