Unraveling the Story of NGF-mediated Sensitization of Nociceptive Sensory Neurons: ON or OFF the Trks?
Abstract
Nerve Growth Factor (NGF) is produced by and affects a number of immune and inflammatory cells. As part of the inflammatory response, NGF directly or indirectly alters the sensitivity of small diameter sensory neurons that communicate noxious information. The question remains as to the receptors and intracellular signaling cascades that mediate this sensitizing action of NGF. Although the general consensus is that NGF produces peripheral sensitization by activating TrkA, recent work suggests that p75 also contributes. Thus, both NGF receptors appear to contribute to peripheral sensitization although whether they act independently or together remains to be determined. Furthermore, controversy exists as to the downstream signaling pathways involved in NGF-induced peripheral sensitization.
Introduction
Nerve Growth Factor (NGF) is a critical trophic factor required during development for the growth and survival of sympathetic neurons, sensory neurons, and neurons in the central nervous system (1, 2). The trophic actions of NGF are largely attributed to the activation of a receptor tyrosine kinase (TrkA) that is expressed on peripheral and central neurons. The NGF-TrkA complex in turn activates a number of signaling pathways in the cell and can be internalized and transported to the cell body to alter gene expression (3, 4). Transgenic mice lacking NGF (5) or TrkA (6) have sensory and sympathetic deficiencies and do not survive long after birth. In addition, genetic studies in patients with congenital insensitivity to pain with anhidrosis (CIPA) reveal mutations in TrkA that impair normal development of sensory neurons (7).
In adult animals, however, NGF is not necessary for neuronal survival (8), although it may still have a trophic role in maintaining the homeostasis of neurons or in sprouting after tissue damage (9, 10). Rather, a large body of evidence supports the notion that NGF’s primary function in adults is to mediate the inflammatory and immune response after tissue injury, especially to initiate and maintain hypersensitivity, a hallmark symptom of inflammation (11, 12). This hypersensitivity manifests as increased sensitivity to a noxious stimulus (hyperalgesia) and/or the perception of a non-noxious stimuli as noxious (allodynia). One mechanism to account for this hypersensitivity is a reduced threshold for firing and an increase in the excitability of small diameter sensory neurons that communicate noxious information to the central nervous system. This phenomenon, termed peripheral sensitization, increases the firing of small diameter sensory neurons and results in an increase in transmitter release from peripheral and central terminals of these neurons (Figure 1⇓). The increased release of the peptide transmitters, substance P (SP) and calcitonin-gene related peptide (CGRP) from peripheral endings of sensory neurons contributes to neuro-genic inflammation (13), whereas increased release of transmitters such as SP and glutamate from endings in the dorsal spinal cord augments nociception (14). Sustained release of transmitters from central terminals also can augment synaptic function between sensory nerve endings and neurons in the dorsal spinal cord (15). This phenomenon, known as central sensitization, also contributes to hyperalgesia and allodynia and may be a major mechanism for chronic pain syndromes.
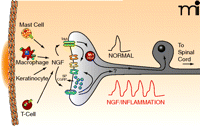
Release of NGF produces sensitization of the peripheral terminals of sensory neurons. Upon localized tissue trauma, mast cells, macrophages, keratinocytes, and T cells all release NGF, which can then interact with its receptors, TrkA and p75, on the nerve endings. Bindings to the NTR activates intracellular signaling cascades that can modulate the activity of different ion channels in the nerve ending or could enhance the release of the neuropeptides, substance P (SP) and/or calcitonin gene-related peptide (CGRP). Under normal conditions, a noxious stimuli might elicit a single action potential whereas after treatment with NGF or as a consequence of inflammation, the nerve now give rise to multiple action potentials. Upon binding the neurotrophin, the TrkA-NGF complex may be retrogradely transported back to the cell body where it can modify the expression of multiple genes, e.g., ion channels. It is possible that p75 lacking NGF can be transported back to the cell body where it also serves to alter gene expression. See text for details.
The ability of NGF to augment nociceptive responses has received much attention and has raised the idea that attenuating the actions of this neurotrophin might prove to be a novel therapy for the treatment of chronic pain (16, 17). This focus on NGF is based on studies demonstrating that administration of NGF produces hypersensitivity and pain in humans (18–20) and reduces nociceptive threshold in animal models of pain (21–23). There are, however, a number of unresolved issues regarding NGF-induced hypersensitivity that need to be addressed. NGF, like many other trophic factors and cytokines, is pleiotropic (24): it is produced by and acts on a number of different inflammatory and immune cells (11, 25). Because NGF has effects on multiple cell types including (but not limited to) sensory neurons, sympathetic neurons, mast cells, and lymphocytes, use of antagonists to block nociception could result in numerous untoward actions. As a result, it is necessary to ascertain which effects of NGF are mediated by a direct action on sensory neurons to produce sensitization and which actions are indirect. Furthermore, the contribution of the two NGF receptors, the TrkA neurotrophin receptor (NTR) and p75 NTR, to peripheral sensitization and the downstream signaling pathways mediating the sensitizing actions of NGF remain unresolved. As discussed below, NGF through activation of its receptors can initiate a variety of intracellular signaling that results in post-translational modification of proteins that contribute to acute sensitization. It also can signal alterations in the expression of proteins that contribute to enhanced excitability of sensory neurons. We examine the evidence that NGF produces peripheral sensitization through direct actions on sensory neurons and also examine the putative signaling pathways that mediate the ability of NGF to produce peripheral sensitization that can lead to enhanced nociception. For discussion of signaling pathways that mediate the other actions of NGF, readers are referred to a number of reviews (26–31).
Nerve Growth Factor Mediates Peripheral Sensitization
During an episode of inflammation, the amount of NGF released into tissues and circulation increases dramatically. In animal models of inflammation, amounts of produced NGF increase in response to administration of Complete Freund’s adjuvant (CFA) (22), carrageenan (32) or airway allergens (33). In humans, increased amounts of NGF are observed in serum from asthmatic patients (34), in tissue biopsies from patients with inflammatory bowel disease (35); in seminal plasma of patients with prostatitis/chronic pelvic pain syndrome (36), in keratinocytes of psoriatic lesions (37), and in synovial fluid from patients with inflammatory joint disease (38, 39).
Measuring behavioral responses to noxious stimuli, Lewin and co-workers first observed that acute systemic administration of NGF causes hyperalgesia to a thermal stimulus within fifteen minutes of injection (40). Other investigators also showed thermal hypersensitivity after acute systemic administration of NGF (41) and after injection directly into the hind paw (42). In contrast, hyperalgesia to a mechanical stimulus has a delayed onset (hours) after injection of NGF, but is consistently observed with chronic exposure to NGF (40). Transgenic mice overexpressing NGF show an enhanced sensitivity in response to mechanical stimuli compared to that observed in wild-type animals, whereas mice with reduced expression of NGF are less responsive to noxious stimuli (43).
Measuring neuronal activity in the rat skin-nerve preparation, Rueff and Mendell observed that acute exposure of the skin to NGF decreased the threshold for firing of small diameter sensory neurons in response to heat (44). This effect of NGF was not observed in skin taken from animals pretreated for four days with the mast-cell granule-depleting drug 48/80, suggesting that the effects of NGF were mediated by release of substances from mast cells. Acute exposure of the receptive fields of primary afferents in the urinary bladder to NGF also resulted in increased excitability of small diameter sensory neurons (45). These data support the notion that acute NGF sensitizes both cutaneous and visceral sensory nerve endings, but whether the increased excitability is caused by a direct action of NGF in the nerves was not determined.
A causal relationship between the increase in NGF amounts and hypersensitivity during inflammation is established by experiments showing that infusion of a neutralizing antibody to NGF reduces withdrawal behaviors in response to noxious thermal and mechanical stimuli (46) and prevents mechanical and thermal hyperalgesia secondary to local injection of CFA into the rat hind paw (21, 22). In an analogous manner, injection of fusion protein TrkA-Immunoglobulin G (IgG)—which binds and sequesters NGF thereby minimizing its ability to bind to its native receptors—diminishes behavioral responses to noxious stimuli and prevents thermal hyperalgesia after local injection of carrageenan (47).
Interfering with the ability of NGF to bind to its receptors also attenuates excitability of sensory neurons after inflammation. In the skin-nerve preparation obtained from rats that received a subcutaneous injection of carrageenan three hours prior to harvesting the tissue, there was an increase in the number of small diameter sensory neurons exhibiting spontaneous firing (48). There also was an increase in sensitivity of sensory neurons to heat and to bradykinin but not to mechanical stimuli. These sensitizing effects of inflammation were abolished by coadministration of a TrkA-IgG fusion protein with the carrageenan (48). In an analogous manner, injection of another TrkA-fusion protein (trkAIg2) into the hind paw of guinea pigs prevented both the increase in spontaneous activity and the decrease in action potential duration induced by intradermal injection of CFA (49). These studies clearly show that NGF released during experimental inflammation is a causative agent in peripheral sensitization, but they do not establish the site of NGF action or the receptors that mediate the action.
NGF-Dependent Gene Expression Contributes To Peripheral Sensitization
Based on the rapid onset of NGF-induced thermal hyperalgesia and the slow onset of mechanical hyperalgesia, Lewin and co-workers proposed that two separate mechanisms mediated acute thermal sensitivity and delayed mechanical sensitivity (40). Indeed the acute effects on thermal stimuli could be explained by activation of signaling pathways in sensory nerve terminals by NGF binding to either of its receptors resulting in phosphorylation of channel proteins that contribute to thermal sensitivity. For example, much evidence demonstrates that acute administration of NGF augments transient receptor potential vanilloid 1 receptor (TRPV1)-mediated currents through post-translational modifications. This ligand-gated ion channel is activated by noxious thermal stimuli and by the algogenic (i.e., pain-producing) agent capsaicin (50), and thus NGF-induced increase in activity of this channel could account for thermal hypersensitivity. The fact that NGF-induced thermal sensitivity is maintained with chronic administration of the neurotrophin (42) suggests that the acute actions of NGF do not decrease with continued exposure to the neurotrophin. This is supported by the observations that acute administration of anti-NGF reverses thermal hyperalgesia induced by inflammation (22) and that sensory neurons grown in culture in the presence of NGF still are sensitized by acute administration of the neurotrophin (51).
The maintenance of NGF-induced hypersensitivity and the late onset of NGF-induced mechanical hyperalgesia also could result from alterations in expression of pro-nociceptive neurotransmitters such as SP or CGRP and/or an increased expression of ion channels that regulate excitability. Indeed, NGF regulates the expression of SP during development (52, 53). Furthermore, although NGF is not required for survival of adult sensory neurons, it still regulates expression of neuropeptides in these cells. Sensory neurons isolated from neonatal and adult rats and grown in culture in the presence of NGF exhibit increased expression of SP and CGRP compared to that observed in cells grown in the absence of NGF (54, 55). This trophic effect on peptide expression appears selective for NGF because neither brain-derived neurotrophic factor (BDNF) nor neurotrophin-3 (NT-3) affected the peptide content of adult sensory neurons in culture (56). Furthermore, chronic administration of NGF significantly augmented the electrical- or capsaicin-stimulated release of substance P from spinal cord slices (57, 58). It was not determined whether this increase in release was secondary to the ability of NGF to increase peptide content in sensory neurons after chronic treatment.
Inflammation induced by intradermal injection of CFA or carregeenan also augments peptide content in sensory neurons (22, 59, 60) and increases release of SP and CGRP from sensory nerve endings in the spinal cord (61, 62). The increase in peptide content after inflammation correlates with the development of hyperalgesia and with an increase in NGF content (22, 59). Furthermore, neutralization of NGF by injection of NGF antibodies not only reduced hypersensitivity but prevented the increase in peptide content suggesting that the inflammation-induced increase in content could account for the NGF-induced hypersensitivity (22, 59).
Long-term exposure to NGF also augments the expression of a number of ion channels that contribute to the excitability of sensory neurons. Indeed, the ability of capsaicin to excite sensory neurons is, in part, dependent on NGF. Sensory neurons isolated from adult rats grown in culture without NGF show a reduced sensitivity to capsaicin compared to cells grown in the presence of NGF (63–66). The ability of NGF to increase capsaicin sensitivity is likely caused by an acute effect of NGF on the TRPV1 channel as well as on the ability of NGF to increase the expression of the TRPV1 channel. Both inflammation and long-term exposure to NGF increase the expression of TRPV1 in sensory neurons (66–69) and this increase is blocked by administration of NGF-specific antibodies (68). The ability of NGF to increase the expression of TRPV1 mRNA is blocked by the receptor tyrosine kinase inhibitor K252A (66), by expression of a dominant-negative Ras, and by a mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitor (67). These data suggest that the ability of NGF to increase TRPV1 expression is mediated by activation of TrkA.
Chronic exposure to NGF also increases sodium channel expression in sensory neurons (70, 71), and this could augment excitability in response to a noxious stimulus. The neurotrophin also increases bradykinin receptor binding (72), expression of P2X3 channels (73), and expression of acid sensing ion channels (74) in sensory neurons. Thus, these NGF-induced increases in expression can render sensory neurons more sensitive to established inflammatory mediators such as bradykinin, ATP, and/or protons (75). Taken together, the data reviewed above support the notion that an NGF-induced alteration in protein expression is one mechanism that could maintain sensory neurons in a state of hypersensitivity such that their threshold for activation is reduced and thus more responsive to mildly noxious stimuli.
Cells Involved In Inflammation That Mediate NGF-Induced Peripheral Sensitization
One unresolved issue regarding the ability of NGF to produce peripheral sensitization is the contribution of various immune and inflammatory cells to the phenomenon. NGF does effect sensory neurons directly; however, when performing in situ experiments or in vitro studies using tissues, it is important to consider the effects of NGF on other cells (Figure 1⇑). NGF is produced by mast cells (76), glial cells (77), keratinocytes (37), fibroblasts (78, 79), lymphocytes (80), and macrophages (81). Many of the cells that are sources for NGF in inflammatory disorders also express the receptors for NGF (82–84). A subset of small diameter sensory neurons co-express TrkA and neuropeptides (85, 86), suggesting that activation of TrkA mediates NGF-induced sensitization via release of these neuropeptides. Of interest, however, are the observations that a majority of small-diameter sensory neurons expressing TrkA also co-express p75 (87, 88). In addition, p75 is found in a subset of small diameter sensory neurons that do not express TrkA. Thus, the distribution of this receptor supports its potential importance in peripheral sensitization.
Localization of NGF receptors on sensory neurons suggests, but does not prove, that NGF acts directly on these neurons to produce hypersensitivity. For example, NGF degranulates mast cells (83, 89) and this effect must be mediated through activation of TrkA as there are no p75 in mast cells (83). This degranulation releases a number of inflammatory mediators that contribute to peripheral sensitization including 5-hydroxytryptamine, trypase, and prostaglandins (14). In addition, because NGF is synthesized and stored in mast cells, the neurotrophin can stimulate its own release from these cells. NGF also contributes to activation and maintenance of T and B lymphocytes, macrophages, and other lymphocytes (90). The T cells and macrophages in turn release cytokines that contribute to chronic pain by altering sensitivity of sensory neurons (23).
Thus, the question remains whether NGF-induced peripheral sensitization is mediated by a direct action of the trophin on sensory neurons or is secondary to NGF actions on other inflammatory or immune cells which in turn release substances that alter the sensitivity of sensory neurons. Pretreating animals with 48/80 attenuates the ability of acutely administered NGF to augment thermal sensitivity, suggesting that mast cells contribute to the action of the neurotrophin (21, 42). Surprisingly, the loss of mast cell activity does not prevent NGF from inducing thermal hyperalgesia hours after administration (21), supporting the notion that different mechanisms mediate the actions of NGF over time. Depletion of mast cells also significantly reduces the increase in NGF levels and mechanical hyperalgesia caused by CFA-induced inflammation (91). Sympathetic nerve endings may contribute to NGF-induced hypersensitivity as sympathectomy by either surgical denervation or chronic exposure to guanethidine significantly attenuates the thermal hyperalgesia produced by intraplantar injection of NGF (91, 92) or attenuates the increased thermal and mechanical sensitivity that is secondary to CFA-induced inflammation (91). As with mast cell granule-depletion, sympathectomy delays the onset but does not abolish CFA-induced hyperalgesia, suggesting that sympathetic nerve endings contribute to acute sensitization but that other mechanisms mediate long-term sensitization. The effects of other cell types on NGF-induced peripheral sensitization have not been examined, although it is interesting to speculate that macrophages and lymphocytes may well contribute to NGF-induced hyperalgesia. These cells produce a number of cytokines that can increase the production of NGF (13–95). Furthermore, cytokines such as Tumor Necrosis Factor–α (TNFα) and Interleukin-1β (IL-1β) contribute to peripheral sensitization and could thereby impact the actions of NGF (23).
Direct actions of NGF on sensory neurons
Although NGF acts on a number of cells that participate in inflammatory and immune responses, much evidence has accumulated showing direct actions of the neurotrophin on sensory neurons. In patch-clamp recordings from small diameter sensory neurons isolated from young adult rats, treatment with either NGF or NT-4/5 augmented the amplitude of the membrane current evoked by capsaicin (96). The finding that NGF can sensitize the neuronal response to capsaicin has since been replicated in many studies using different model systems. For example, TRPV1 expression in the heterologous systems of Xenopus oocytes and HEK cells showed that the thermal sensitivity to a heat stimulus was increased after exposure to NGF (97). Similar results were reported for the increased heat sensitivity of isolated small diameter sensory neurons that coexpressed TrkA (98). In addition to sensitization of a ligand-gated current, neuronal excitability is also enhanced by acute exposure to NGF wherein the number of action potentials generated by a ramp of depolarizing current in adult sensory neurons grown in culture was increased by NGF (51). Analogous to the enhancement of TRPV1 currents, acute exposure to NGF increased the capsaicin-evoked increase in the levels of intracellular free Ca2+ in small-diameter sensory neurons isolated from neonatal rats [postnatal day two (P2)-P5] (99). These results are consistent with previous studies wherein NGF augmented the capsaicin-gated current in adult sensory neurons (69, 96, 100). In these neonatal neurons, Bonnington and McNaughton found that treatment with NGF had no effects on the resting levels of intracellular Ca2+ (99). Of interest, the increase in intracellular free Ca2+ produced by a high concentration of extracellular potassium was not affected by NGF, suggesting that the sensitizing effect was limited to stimuli that activate the TRPV1 channel. This observation is inconsistent with the finding that the number of action potentials evoked by depolarizing current was increased by NGF (51). Additionally, a lack of effect of NGF on resting Ca2+ is in contrast to the report by Lamb and Bielefeldt who showed that both NGF and BDNF elevated intracellular Ca2+ in visceral sensory afferents isolated from the nodose ganglia (101). Furthermore, Zhu et al. failed to observe an increase in the capsaicin-gated current after treatment with NGF in sensory neurons isolated from neonatal rats (102). These authors also found that the period between P4 and P10 was critical for the development of NGF-mediated sensitization even though these neonatal neurons were sensitized by bradykinin (102). It is not at all clear why such differences are observed in what should be very similar model preparations. These striking differences in the results obtained with neonatal sensory neurons suggest that sensory neurons at this developmental stage may not be a reliably consistent model preparation in which to determine the exact signaling mechanisms regulating cellular excitability.
The Neurotrophin Receptors: p75 and TrkA
NGF binds and activates two distinct receptors, the p75 and TrkA neurotrophin receptors (NTR)s. Classically, these receptors were distinguished by their binding affinity for NGF and their rates of NGF dissociation (103). NGF binds to a low affinity site (Kd ~10−9 M) as well as to a high affinity site (Kd ~10−11 M). Further studies demonstrate that the low affinity site was attributed to a protein having a molecular mass of ~80 kDa, designated p75, whereas the high affinity site was a protein having a molecular weight of ~140 kDa, designated TrkA. The low affinity site is characterized by a rapid rate of dissociation of NGF (t1/2 ~3 sec), whereas dissociation from the high affinity site is much slower (t1/2 ~10 min). Despite the differences in affinity, NGF concentrations in tissues after inflammation or injury as well as after exogenous administration likely are sufficiently high to activate both receptors.
The p75 NTR, a member of the TNF receptor (TNFR) superfamily, is composed of an extracellular NGF-binding domain that contains the four cysteine-rich motifs that are characteristic of the TNFR family (Figure 2⇓). There is a short transmembrane spanning domain and a short intracellular domain that is not associated with any inherent catalytic activity but does contain a FAS “death domain” also characteristic of the TNFR family. The cytoplasmic tail has sites for binding of various proteins associated with p75 signaling (Figure 2⇓). The p75 NTR has been referred to as a “common” or “pan” NTR because of its ability to bind all neurotrophins (i.e., NGF, BDNF, NT-4/5, and NT-3) with equal affinities although the rates of association and dissociation may vary with the particular neurotrophin.
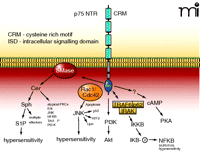
The p75 NTR and its associated intracellular signalling pathways. The p75 NTR is made up of four extracellular cysteine rich motifs (CRM), a single membrane spanning domain, and an intracellular signaling domain (ISD). NGF binding causes the activation of sphingomyelinases (SMase) that lead to the liberation of ceramide (Cer) and subsequently to sphingosine (Sph) and sphingosine 1-phosphate (S1P). Ceramide and sphingosine can activate a multitude of effector pathways. In small-diameter sensory neurons the generation of S1P appears to enhance the firing of action potential through the modulation of a TTX-resistant sodium current and the suppression of a delayed rectifier-like current in sensory neurons. The small G proteins Rac1/Cdc42 associate with the ISD, which results in the activation of JNK. In many cell systems, activation of JNK is critically involved in apoptosis, however, in sensory neurons, activation of JNK gives rise to hypersensitivity. The ISD, through an undefined mechanism, results in the activation of PI3K with downstream activation of Akt. The ISD can associate with the complex formed by TRAF-IRAK-p62 to activate IKKβ, which then phosphorylates IκB and release NF-κB. In sensory neurons, activation of the NF-κB pathway appears to be anti-nociceptive. Lastly, activation of p75 somehow leads to increased levels of intracellular cyclic AMP and activation of PKA. See text for details
The TrkA NTR shares a significant homology with the proto-oncogene tropomyosin receptor kinase (trk) and TrkA is localized in cells that require NGF for their survival (103, 104). The extracellular domain of TrkA (Figure 3⇓) is composed of two cysteine-rich motifs that are separated by three leucine-rich motifs; these are followed by two Ig-like motifs. These Ig-like motifs are found in other members of the receptor tyrosine kinase family such as platelet-derived growth factor receptor (PDGFR) and epithelial growth factor receptor (EGFR). There is a single short transmembrane spanning region that is followed by the juxtamembrane region. Beyond the juxtamembrane region is the catalytic domain that contains the conserved tyrosine residues whose phosphorylation is necessary for the binding of different adaptor molecules as well as effectors of TrkA signaling. Like other members of the receptor tyrosine kinase family, TrkA is characterized by its dimerization upon binding by NGF.
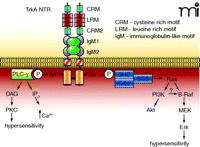
The TrkA NTR and its associated intracellular signaling pathways. The TrkA NTR is made up of multiple extracellular domains that consist of two cysteine-rich motifs (CRM), three leucine rich motifs (LRM) and two immunoglobulin-like motifs (IgM). Binding of NGF results in dimerization and the cross-phosphorylation of tyrosine residues on the intracellular signalling domains. These tyrosines form the binding sites for the scaffolding proteins Shc-Grb2-SOS, which leads to the activation of the small G-protein Ras, followed by activation of the serine-threonine kinases Raf, MEK, and the MAPK, Erk. In sensory neurons, the activation of Erk has been linked to hypersensitivity. Ras can also activate PI3K and its downstream effector Akt. In some cells, PI3K can activate B-Raf which leads to phosphorylation of Erk. The phosphorylated tyrosines also form the binding site for PLC-γ, which consequently results in the activation of PKC. It is well established that PKC plays an important role in regulating the sensitivity of sensory neurons to noxious stimulation.
Although a majority of the actions of NGF are attributed to activation of either TrkA or p75, growing evidence suggests significant cross-talk between these receptors. Indeed, p75 can augment binding affinity of TrkA. In experiments where TrkA cDNA is expressed in 3T3 cells, only low-affinity binding (~10−9 M) was observed. In a fibroblast cell line (PAEC1 cells) expressing the cDNA for p75, only low-affinity binding was detected. Surprisingly, when membranes from these two cells were fused, both low and high affinity sites were detected (105). In vivo studies also support the idea that p75 enhances the sensitivity of TrkA. In p75 knock-out mouse (106), the EC50 for the ability of NGF to promote neuronal survival of embryonic trigeminal sensory neurons was three- to four-fold higher than in the heterozygous mouse (107). In addition, when the interaction between p75 and TrkA was limited by either using a p75-blocking antibody or by decreasing the expression of p75, the corresponding activation of TrkA by NGF was reduced (108, 109). Studies also suggest that p75 contributes to TrkA-mediated survival and differentiation in neuroblastoma cells (110) and that it enhances TrkA-mediated phosphorylation of the adapter protein Shc (111). These results suggest that p75 plays a critical role in maintaining the high affinity binding of NGF to TrkA and that it may augment downstream signaling by TrkA (112).
The p75 NTR may not be unique in its capacity to enhance the sensitivity of TrkA for NGF. A protein called neurotrophic receptor homologue 2 (NRH2) has a great deal of similarity to p75 (113). NRH2 has a truncated extracellular domain that is incapable of binding NGF, but the transmembrane and cytoplasmic domains are very similar to p75. Also, like p75, NHR2 enhances the binding affinity of TrkA for NGF. NRH2 has been detected in a large portion of neonatal rat sensory neurons (P2/3); however, the role of NHR2 in NTR activation and signaling efficacy has yet to be determined.
Signaling pathways activated by neurotrophin receptors
The p75 NTR
Early work suggested that p75 had no catalytic activity and thereby did not activate any downstream signaling pathways. More recent work, however, has demonstrated this not to be the case, and that activation of p75 by NGF, as well as by its other ligands such as BDNF, produces significant intracellular signaling events that mediate such processes as cell growth, apoptosis, and modulation of neuronal excitability (28, 31). For brevity, we will focus our discussion on signaling pathways where data supports the notion that they mediate peripheral sensitization. The initial studies that demonstrated a p75-mediated signaling pathway independent of TrkA activity showed that NGF produced an increase in the levels of intracellular ceramide and a corresponding decrease in the content of sphingomyelin in NIH 3T3 cells (114). Indeed, NGF binding to p75 activates a neutral sphingomyelinase that metabolizes membrane sphingomyelins that then liberates ceramide (Figure 2⇑). These findings suggest some functional signaling similarity between p75 and TNFR1, which also is capable of activating the sphingomyelin signaling cascade.
Ceramide can be further metabolized to sphingosine by an enzyme called ceramidase, and sphingosine can be phosphorylated by sphingosine kinase to produce sphingosine 1-phosphate (115, 116). Ceramide also can be metabolized by ceramide kinase to ceramide-1-phosphate. Thus, activation of the sphingomyelin cascade by NGF can give rise to at least four intracellular signaling molecules that may contribute directly or indirectly to the inflammatory response (116). Acute exposure of isolated sensory neurons to ceramide or to sphingosine 1-phosphate increases the number of action potentials generated by a ramp of depolarizing current, augments tetrodotoxin (TTX)-resistant sodium currents, and inhibits potassium currents in isolated sensory neurons (51, 117). Liberation of ceramide can lead to the activation of mitogen-activated protein kinases (MAPKs) and NF-κB depending on the cell system, and these molecules have been implicated in peripheral sensitization and enhanced pain perception. Ceramide-1-phosphate also increases phospholipase A2 (PLA2) activity, whereas sphingosine 1-phosphate (presumably by activation of NF-κB) increases the expression of cyclooxygenase-2 (118). These actions can augment the production of prostaglandins which may contribute to peripheral sensitization.
NGF binding to p75 also activates other signaling pathways associated with sensitization of sensory neurons (Figure 2⇑). This association suggests potential mechanisms for the involvement of p75 in NGF-induced hyperalgesia, but to date few studies have addressed this notion. In the absence of an interaction with TrkA, activation of p75 initiates apoptosis in a variety of cells (119). One signaling pathway involved in this effect is the activation of small G-proteins that leads to the activation of c-jun N-terminal kinase (JNK), which, in turn, activates various downstream apoptotic pathways such as the transcription factors c-jun, ATF2, and p53 (119). Activation of JNK also may contribute to hypersensitivity after inflammation or nerve injury. Inflammation of the rat hind paw by injection of CFA or administration of NGF produces thermal hypersensitivity associated with an increase in the levels of phosphorylated JNK in the L5 ganglion (120). Inhibition of JNK activity reduces the extent of the thermal hypersensitivity produced by either inflammatory agent. Using the neuropathic pain model involving ligation of the L5 spinal nerve, immunohistochemical detection of phosphorylated JNK was dramatically increased in both the small and medium diameter sensory neurons (121, 122). Pretreatment with an inhibitor of JNK activity blocked mechanically induced allodynia resulting from nerve injury. It is unknown whether p75, TrkA, or both mediate the activation of JNK to give rise to hypersensitivity in these experimental models. This remains an important question for future studies.
The p75 NTR associates with the tumor necrosis receptor–associated factor 6 (TRAF6) and forms a scaffolding platform for the subsequent binding of the IL-1 receptor associated kinase (IRAK) (123), which is a serine-threonine kinase involved in activation of NF-κB. The p75-TRAF6-IRAK forms a complex with atypical PKC–interacting protein (also termed p62). This complex ultimately associates with IκB kinase (IKK), which phosphorylates IκB and releases NF-κB (124). In addition, TRAF proteins can form complexes with receptor-interacting protein 2 (RIP2), which is thought to be involved in the activation of NF-κB (125, 126). Activation of NF-κB through p75 appears important in neuronal survival after injury (127). A blocking antibody to p75 completely suppressed NGF-mediated activation of NF-κB and greatly diminished the survival of embryonic trigeminal neurons (128). NGF, through activation of p75, also increases NF-κB translocation to the nucleus in Schwann cells (127). Exogenous administration of a single dose of NGF into the rat paw does not increase NF-κB activity in nuclear extracts from the lumbar dorsal root ganglia (129). Inhibitors of IKK or NF-κB decoy molecules attenuate hyperalgesia produced by inflammation (130, 131) and by injury to the sciatic nerve (130, 132). In light of this work, further studies seem warranted to determine whether the p75-induced activation of NF-κB could directly or indirectly mediate hypersensitivity of sensory neurons after inflammation.
In a variety of cell types, p75 also promotes cell survival through its activation of Akt [also called protein kinase B (PKB)], which is dependent on the activation of phosphoinositide 3′-kinase (PI3K) (133, 134). This effect appears to be independent of TrkA, despite the fact that activation of TrkA also promotes survival through the PI3K-Akt pathway. This idea is supported by the observation that treatment of PC12 cells with antisense oligonucleotides targeted to p75 resulted in decreased levels of phosphorylated Akt produced by NGF (111). These observations raise an interesting question regarding the presumed activation of PI3K solely by TrkA in the sensitization of the current conducted by TRPV1. A possible role for activation of PI3K-Akt by p75 in the regulation of sensory neuron sensitivity or excitability has yet to be determined.
In PC-12 cells, exposure to both NGF and BDNF can lead to a rapid elevation in intracellular levels of cyclic AMP (cAMP) (135). This observation suggests that the increase in cAMP is mediated by p75, as PC12 cells do not express the TrkB NTR, and thus the actions of BDNF are likely through p75. Similar observations were reported for isolated mouse cerebellar neurons, which express the p75 but not TrkA, wherein NGF rapidly elevated intracellular cAMP (136). These authors also showed that in isolated hippocampal neurons, treatment with NGF caused an outgrowth of neurites that was blocked by the protein kinase A (PKA) inhibitor KT5720. These results suggest that an NGF-induced increase in PKA activity has a functional role in the growth of neurons. Recently, it was reported that in PC12 cells both cAMP-dependent and NGF signaling pathways produce phosphorylation of Src at Ser17 (137). This phosphorylation of Src leads to the activation of the small G-protein Rap1, which in turn can activate extracellular-regulated protein kinase (Erk, a member of the MAPK family) (138). The NGF-induced activation of Rap1 is blocked by either the non-selective serine-threonine kinase inhibitor H-89 or the Src inhibitor PP2, suggesting that NGF might somehow activate PKA. This potential cross-talk of signal pathways may play a role in NGF-induced sensitization of sensory neurons. If NGF increases cAMP in sensory neurons, the cyclic nucleotide can bind to either exchange proteins that are activated directly by cAMP (Epacs) (139) or to PKA. Epacs are guanine nucleotide exchange factors that increase activity of small G-proteins and downstream MAPKs and have been implicated in the sensitization of sensory neurons (140). Increases in cAMP and activation of PKA mediate peripheral sensitization produced by prostaglandins (141, 142), but the role of Epacs in this signaling pathway in NGF-induced sensitization has not been explored.
The cellular mechanisms mediating the capacity of NGF to increase intracellular cAMP remain to be determined. A recent study, however, has shown that NGF treatment of PC12 cells produces a rapid (<2 min) increase in cAMP; this increase was blocked when cells were pretreated with BAPTA-AM, a Ca2+ chelator (143). Significantly, the NGF-induced increase in cAMP was blocked by KH7, an inhibitor of soluble adenylyl cyclase. Soluble adenylyl cyclase is very different from the conventional transmembrane forms of adenylyl cyclase in that it is activated by increases in bicarbonate or Ca2+ but not by forskolin (144). These results suggest that NGF somehow leads to an increase in intracellular Ca2+, which then activates soluble adenylyl cyclase with subsequent activation of PKA. It is clear that our understanding of the specific mechanisms and signaling pathways whereby NGF stimulates intracellular cAMP, PKA, and their consequent downstream effects is quite limited.
The Trka NTR
In contrast to p75, TrkA-mediated signaling is initiated by the autophosphorylation of critical tyrosine residues in the intracellular domain. These residues then form the binding sites for different adapter/scaffolding proteins that serve as sites for additional effector proteins as well as effector enzymes such as phospholipase C (Figure 3⇑). The major signaling effectors activated by TrkA are the small G-proteins Ras and Rap1, leading to activation of MAPKs, PI3K, and phospholipase C–γ (27, 29–31). Recent work indicates that these pathways can mediate the hypersensitivity of sensory neurons.
The binding of adapter proteins such as Shc, Grb2, and the guanine-nucleotide exchange factor SOS to TrkA leads to the activation of Ras with a subsequent increase in activity of the MAPK pathway composed of Raf, MEK, and Erk. In addition, the adapter protein CRK and the guanine-nucleotide exchange factor C3G activate Rap1, which maintains activation of Erk for an extended period of time (138). Activated Erk phosphorylates several different substrates and can migrate to the nucleus where it activates specific transcription factors. In sensory neurons grown in culture, NGF increases the levels of phosphorylated Erk (67, 69, 145). This increase can be rapid where amounts of phosphorylated Erk are increased within five minutes after NGF treatment or sustained over an extended period of time (145). NGF-induced phosphorylation of Erk is blocked by the PI3K inhibitors LY294002 or wortmannin, suggesting that this kinase activity lies upstream of Erk. As mentioned above, NGF exposure also can activate B-Raf and Ras in these neurons, and the activation of B-Raf is blocked by LY294002, whereas activation of Ras is unaffected (145). In addition, RapGAP1 (a GTPase accelerating protein for Rap) blocks the slow phase but not the rapid phase of Erk phosphorylation. These results indicate that the activation of Ras is upstream from the PI3K modulation of B-Raf activity and that there may be parallel signaling pathways that mediate the activation of Erk. Erk has been implicated in hyperalgesia and in sensitization of sensory neurons. The phosphorylation of Erk is increased in small-diameter sensory neurons by capsaicin and by noxious thermal stimuli (146). Inhibition of Erk phosphorylation by the MEK inhibitor U0126 attenuated capsaicin-evoked thermal hyperalgesia (146) and mechanical hyperalgesia caused by intradermal injection of epinephrine into the rat paw (147). Furthermore, in acutely dissociated sensory neurons, U0126 attenuates the ability of capsaicin to enhance heat-evoked currents (148). These data clearly implicate Erk activation (as indicated by phosphorylation) in sensitization of sensory neurons.
Binding of NGF to TrkA also activates PI3K (independent of Ras activity) by binding to adapter proteins linked to TrkA. In addition to the activation of B-Raf, PI3K catalyzes the phosphorylation of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) at the 3-position; various species of inositol phosphates are involved in the activation of Akt. Akt, in turn, can phosphorylate a number of downstream effectors that can modulate the activity of the transcription factor nuclear factor–κB (NF-κB) and other as yet unknown effectors. Although a number of recent studies suggest that PI3K mediates the sensitivity of sensory neurons, the roles of Akt and its downstream effectors have yet to be determined.
Tyrosine phosphorylation of TrkA leads to the binding and activation of PLC-γ (Figure 3⇑), which liberates the second messengers inositol trisphosphate and diacylglycerol (DAG) from PIP3. Inositol trisphosphate (IP3) binds to its receptor in the endoplasmic reticulum and produces the release of intracellular Ca2+ stores, whereas DAG binds to a number of downstream effectors (149, 150), most notably the classic and novel isoforms of protein kinase C (PKC). Much work has shown that activation of PKCs mediate sensitization of sensory neurons. Phorbol esters, which activate novel and classic PKCs, injected into the hind paw of rats produce a thermal hyperalgesia that is blocked by co-administration of PKC inhibitors (151). A component of this hyperalgesia likely involves sensitization of sensory neurons because phorbol esters or DAG analogs increase capsaicin-induced calcium entry into isolated sensory neurons (152) and increase capsaicin-, heat-, or proton-evoked currents (153, 154). Activators of PKC also increase capsaicin-evoked release of neuropeptides from isolated sensory neurons (155). Although it is well established that activation of PKC is one mechanism to account for sensitization of sensory neurons by the inflammatory mediator bradykinin (152, 156), the role of PKC in NGF-induced sensitization remains controversial.
Peripheral Sensitization
Mediated By NGF Receptors
Although there are multiple signaling pathways activated by either p75 or TrkA that mediate NGF-induced peripheral sensitization, the question remains as to which pathways are causal in this phenomenon. This question is complicated by the fact that NGF can alter sensitivity of sensory neurons acutely by initiating post-translational modifications of ion channels and chronically by altering gene expression. The relative importance of each of these components in sensitization with chronic exposure to NGF either exogenously or secondary to inflammation remains unknown.
Although the general consensus seems to focus on activation of TrkA as mediating the long-term effects of NGF on sensory neurons, the data supporting this notion are mostly correlative. It is well established that the NGF-TrkA complex can be internalized and transported to the cell body (Figure 1⇑) where it activates gene transcription (153, 157). This transport can occur in vesicle-like structures, endosomes, and these can contain NGF, TrkA, and MAPKs (4). It also is likely that NGF signals can be transported without internalization of TrkA (158). In addition to internalization, NGF binding to TrkA activates Ras and other small G-proteins that lead to activation of the MAPKs and to altered gene expression (159). Activation of p75 also can result in alterations of gene expression. For example, increases in bradykinin binding sites in cells treated with NGF are blocked by administration of a p75-specific antibody. However, the capacity of NGF to alter bradykinin binding is lost in transgenic mice with a deletion in exon III of p75 (72). Thus, additional studies are needed to establish the contribution of each NTR subtype to the long-term changes in sensory neurons produced by NGF.
For the acute, sensitizing actions of NGF, it seems unlikely that internalization of the NGF-TrkA complex—with subsequent alterations in gene expression—is involved because of the time required to transport the complex from the periphery to the nucleus. This does not preclude the potential importance of signaling pathways downstream of TrkA with effectors that alter phosphorylation of proteins and enhance excitability of sensory neurons. For example, intradermal injection of the PI3K inhibitor LY294002 or the MEK inhibitor PD98059 attenuates NGF-induced thermal hyperalgesia, suggesting the involvement of TrkA as a mediator of NGF effects (69). These authors suggest that the PI3K activity is upstream of Erk because LY294002 blocks NGF-induced increase in phosphorylation of Erk. It is important to note, however, that activation of PI3K also occurs with NGF binding to p75. Reducing the expression of TrkA in the saphenous nerve, by intrathecal administration of antisense oligodeoxynucleotides (AS-ODN) specific for TrkA, abolishes NGF-induced mechanical hyperalgesia (160), suggesting TrkA mediates NGF-induced hypersensitivity. In a neuropathic pain model, however, intrathecal injection of p75 AS-ODN reduced expression of this receptor in the dorsal root ganglia (DRG) and attenuated thermal hyperalgesia (161). p75 AS-ODN administration also prevents the injury-induced increase in phosphorylation of p38 (a MAPK) and TrkA. Intrathecal injection of the receptor tyrosine kinase inhibitor K252A also blocks phosphorylation of both TrkA and p38 (161) leading the authors to suggest that an interaction between the two NTRs mediates hypersensitivity after nerve injury.
TrkA might mediate NGF-induced hyperalgesia, as transgenic mice that have a deletion in exon III of p75 gene (106) exhibit a significant elevation in basal nociceptive thresholds and fewer numbers of all neuronal types in the DRG as compared to the numbers and thresholds observed in DRGs from wild-type mice (162). Despite the elevated threshold for nociception, which is likely caused by a reduction in the number of nociceptors in the animal, systemic administration of NGF still induces mechanical hyperalgesia after four hours (163). As pointed out by Bergmann et al., however, it is not clear whether this hyperalgesia is mediated by a direct action of NGF on sensory neurons or indirectly through its actions on other inflammatory cells. Because mast cells express only TrkA (83), the NGF-induced degranulation and release of inflammatory mediators would still occur in p75 knock-out animals and this could produce hyperalgesia. In addition, further examination of the studies with p75 exon III−/− mice show that they still express a splice variant of p75 (164). There is now a complete p75 knock-out animal with a deletion of p75 exon IV and this animal shows an exaggerated loss of peripheral nerves compared to p75 exon III knock-out mice (164). To date, however, studies in the exon IV knock-out mouse have not been performed to examine the important question regarding the ability of NGF to produce hyperalgesia and/or to sensitize sensory neurons.
In isolated sensory neurons, measurements of excitability have not resolved the question of which NTR mediates sensitization. In their initial studies, Shu and Mendell were able to block the ability of NGF to augment the amplitude of the capsaicin-evoked current with the receptor tyrosine kinase inhibitor K252A (96), suggesting that the effects of NGF were mediated by TrkA. However, the selectivity of this compound to receptor tyrosine kinases is questionable because K252A also inhibits a number of other protein kinases such as PKC, PKA, and cGMP-dependent protein kinase (PKG) (165, 166). The involvement of TrkA in NGF-induced heat sensitivity is supported by the fact that sensory neurons sensitized by NGF expressed TrkA, whereas those neurons that were not sensitized by NGF were found to be TrkA negative (98). Measurements of p75 expression were not performed in this study. In oocytes and human embryonic kidney (HEK) cells coexpressing TrkA and TRPV1, NGF augments the capsaicin-gated currents, and this effect is not altered by the coexpression of both TrkA and p75 (97).
The p75 NTR also might directly mediate sensitization of sensory neurons. Patch clamp recordings from NGF-treated, capsaicin-sensitive small-diameter sensory neurons isolated from young adult rats indicated a significant increase in the number of action potentials evoked by a ramp of depolarizing current (51). NGF enhanced the TTX-resistant sodium current and suppressed a delayed rectifier-like potassium current, suggesting that modulation of these membrane currents contributed to the increased excitability produced by NGF. Indeed, when sensory neurons were treated with a blocking antibody for p75, the sensitizing action of NGF to increase the number of action potentials was inhibited (167). Consistent with the inability of NGF to augment the number of action potentials, the administration of p75 blocking antibody also prevented suppression of the potassium current by NGF.
NGF-Induced Signaling Cascades
Controversies also exist as to the downstream signaling pathways that mediate the ability of NGF to alter the excitability of isolated sensory neurons. Indeed, results of studies with a similar design have been surprisingly inconsistent. In a follow-up study to their original work, Shu and Mendell observed that pretreating sensory neurons with PKA inhibitors H89 or PKI14–22, suppressed the NGF-induced enhancement of the capsaicin-gated current recorded from adult sensory neurons (100). In contrast, in neonatal sensory neurons, the inhibition of PKA by another drug, KT5720, had no effect on the capacity of NGF to augment the increase in intracellular Ca2+ concentration evoked by capsaicin (99). The capsaicin-evoked elevation in intracellular Ca2+ is thought to reflect increased TRPV1 activity, although capsaicin also releases Ca2+ from intracellular stores (168). Similar to the role of PKA, inhibitors of PKC have exhibited variable results; in one study both staurosporine and BIM reduced the sensitization by NGF (99) whereas others report that staurosporine, Ro-31-8425, or BIM had no effect on the NGF-induced sensitization of the capsaicin-evoked current (97, 100).
The involvement of PLC-γ in NGF-induced sensitization is also controversial. In the HEK heterologous expression system, TrkA, TRPV1, and PLC-γ are thought to form a signaling complex that then results in the enhancement of TRPV1 activity by NGF (97) because a mutant TrkA incapable of coupling to PLC–γ fails to enhance capsaicin-evoked current when exposed to NGF. Furthermore, NGF-induced sensitization of small-diameter sensory neurons to heat is suppressed by the PLC inhibitor U73122 (98). In contrast, neomycin, an inhibitor of PLC-γ had no effect on the NGF-induced sensitization of the increase in Ca2+ concentration evoked by capsaicin in neonatal sensory neurons, whereas treatment with the other PLC inhibitor, U73122, elevated levels of resting calcium (99).
Based on the ability of NGF to activate Ras (and consequently Erk) in many model systems, it is surprising that MEK inhibitors have little effect on the ability of NGF to augment either capsaicin-evoked currents (97, 100) or increases in intracellular Ca2+ (99). This contrasts the recent work of Zhuang and co-workers who show that inhibition of Erk reduces the ability of NGF to augment capsaisin-evoked currents (69). Inhibition of PI3K by either wortmannin or LY294002 also blocks the NGF-induced sensitization of sensory neurons in two different preparations (69, 99). Taken together, these studies demonstrate that NGF, through the activation of various signaling cascades, can modulate the activity of TRPV1, but the specific signaling pathways involved are not clear. A recent study by Zhang et al. suggests another possible mechanism for the NGF-induced enhancement of TRPV1 activity (169). Using a heterologous expression system, these authors found that exposure to NGF appeared to initiate a rapid insertion of additional TRPV1 channels into the plasma membrane, which can account for an increase in intracellular calcium (169). This NGF-mediated sensitization of the capsaicin-evoked increase in intracellular Ca2+ levels was blocked by the Src inhibitor PP2. Additional experiments showed that Src phosphorylated Y200 of TRPV1, and that this phosphorylation event was critical for the insertion of TRPV1 into the membrane.
Recent work in our laboratories has focused on the sphingolipid signaling pathway as causal in mediating NGF-induced sensitization of sensory neurons. As mentioned above, NGF through p75 activates neutral sphingomyelinase, which in turn increases hydrolysis of membrane sphingomyelin to liberate ceramide (114). Exposing dissociated adult sensory neurons to C2-ceramide, a membrane-permeable analog of ceramide, increases the action potential–firing evoked by depolarizing current in a manner analogous to that observed with acute NGF treatment (51). Furthermore, inhibition of the neutral form of sphingomyelinase with glutathione (70, 171) blocked the capacity of NGF but not ceramide to augment the number of action potentials evoked by the ramp (51). Internal perfusion of small-diameter sensory neurons with either sphingosine or sphingosine 1-phosphate via the patch clamp recording pipette also produced an increase in the number of evoked action potentials with a time course that was very similar to that observed with NGF perfusion (117). Inhibition of sphingosine kinase by dimethylsphin-gosine blocked the sensitizing effect of both NGF and internally perfused sphingosine but did not affect the actions of sphingosine 1-phosphate (117). Taken together, these results indicate that NGF, via activation of p75, can liberate ceramide. This sphingolipid is then metabolized to sphingosine 1-phosphate which through presently unknown mechanisms, leads to the modulation of ion channels and ultimately the enhancement of neuronal excitability. At present, it is not known whether TrkA makes any contribution to the NGF-mediated signaling via p75. Also, the effector systems whereby sphingosine 1-phosphate might alter the activity of different ion channels are unknown. These are two important areas for future investigation that will lead to a better understanding of exactly how these signaling pathways lead to increased excitability.
Interactions Between the TrkA and p75 Signaling Cascades
There are several observations concerning the interaction between the p75 and TrkA NTRs with potential relevance to the NGF-mediated sensitization of sensory neurons. NGF binding to TrkA appears to suppress the capacity of p75 to activate sphingomyelinase (172). In PC12 cells, which express both p75 and TrkA, exposure to NGF failed to elicit the hydrolysis of sphingomyelin. After pretreatment with K252A, the ability of NGF to cause hydrolysis of sphingomyelin was restored, suggesting that the tyrosine kinase activity of TrkA somehow affected the ability of p75 to activate sphingomyelinase. This finding is contrary to two more recent observations. Pretreatment of PC12 cells with BNDF, which activates p75 in these cells, decreased the levels of tyrosine phosphorylation of TrkA produced by the TrkA-specific ligand NGF3T (173). C2 ceramide exposure also decreased the NGF3T-mediated phosphorylation of TrkA. These results suggest that activation of the p75 can negatively impact the activation of TrkA. In addition, the ability of TrkA to suppress sphingomyelinase is difficult to reconcile with our observations (51, 117) wherein NGF augments the excitability of small-diameter sensory neurons through a sphingomyelinase-dependent pathway. Along these same lines, TrkA appears to suppress the ability of the p75 pathway to stimulate NF-κB by somehow modulating the activity of the p75-TRAF6-IRAK-p62 complex, perhaps by association of TrkA with p62 (123, 174). There also may be convergence of activation of TrkA and p75 on downstream signaling. Activation of the TrkA cascade resulted in the rapid and sustained activation of MAPKs, whereas downstream activation of Akt was delayed for approximately thirty minutes (175). Stimulation of the p75 cascade produced a rapid but transient activation of MAPK with no consequent activation of Akt. However, stimulation of both pathways produced a rapid and sustained activation of MAPK with a rapid activation of Akt not seen with activation of either pathway alone. Taken together, these observations suggest that there is a complex interaction between p75 and TrkA and their downstream signaling pathways that we are only beginning to understand.
Musings on mystifying matters
Overall, the experimental observations reviewed above do not provide a clear picture of which receptors or signaling cascades mediate the alterations in the sensitivity of sensory neurons produced by NGF. In these various studies, the results are complicated by the use of different endpoints to measure peripheral sensitization. The most clinically relevant endpoints are the measurement of alterations in behavioral responses to noxious stimuli after NGF exposure. These studies are complicated, however, by the fact that inflammation and exogenous administration of NGF affect a number of different inflammatory and immune cells and these in turn release inflammatory mediators that can sensitize sensory neurons. Thus, in vivo studies attempting to elucidate NGF signaling in sensory neurons must consider the impact of other inflammatory mediators and their respective signaling pathways. Studies utilizing sensory neurons grown in culture clearly provide more meaningful results in assessing the direct actions of NGF on these cells. An additional advantage in these studies is that a number of different endpoints can be measured, including: 1) alterations in membrane currents evoked by noxious stimuli; 2) increases in action potential–firing; 3) increases in calcium entry; and 4) increases in transmitter release. When confronted with a multitude of measurements, however, it is important to consider the possibility that these different endpoints evaluating the state of neuronal sensitivity may involve distinct NTRs and signaling pathways. For example, it is clear that NGF though p75 alters excitability in response to a ramp of depolarizing current (51, 167). In like manner, convincing evidence supports the idea that activated TrkA can alter capsaicin-evoked currents, but different signaling pathways may regulate calcium entry. Given that a number of different factors contribute to the overall sensitivity of sensory neurons, it will be important to ascertain how these components contribute to the functional alterations in neuronal activity and subsequent release of transmitters from these neurons.
Consideration must be given to the age of the animal from which cells are harvested (see above) and the culture conditions used in the various experiments since these factors can impact the results (176). A number of contemporary studies have been performed using expression systems rather than native cells. The obvious advantage of these cell lines is the control of protein expression, and such results are valuable in demonstrating that interactions between NGF and specific signaling pathways may exist (i.e., a proof of concept). These studies are limited, however, because signaling paradigms can be regulated in different manners in these expression systems and signaling outcomes can be modified by overexpression of proteins. Thus, it is critical that additional studies be performed in native cells to determine the potential functional significance of results observed in expression systems.
Another complication is that NGF-induced peripheral sensitization involves both acute and chronic post-translational modifications of proteins as well as chronic alterations in gene expression. In a number of instances, especially in inflammatory models or with repeated injections of NGF, it is difficult to determine if changes in sensitivity are secondary to phosphorylation of effectors that could potentially increase or reduce excitability or to increased production of effectors. Given that different signaling pathways are likely involved in these processes, different results could be obtained based on the time of the measurement after experimental manipulation and/or the duration of inflammation or neurotrophin exposure.
Of greatest significance, however, is the fact that among the many signaling pathways activated by NGF, there is very likely a significant amount of cross-talk between them. Dissecting the exact sequence of events that regulate the activation of these pathways has been limited by the paucity of selective inhibitors to block enzymes in specific pathways. As mentioned above, one indicator commonly used to distinguish between NGF activation of TrkA and p75 is whether the observed effect is blocked by the receptor tyrosine kinase inhibitor K252A. Interpretation of results using this drug are limited by the concentration of drug used, becuase at higher concentrations, K252A also inhibits a number of other kinases. Furthermore, although a blocking antibody to p75 is available, no such antibody currently exists for TrkA. The use of both TrkA and p75 knock-out mice for studies of sensitization also is limited because both receptors participate in the growth and survival of both sympathetic and sensory neurons. Furthermore, a potential interaction between TrkA and p75 suggest that it is difficult to selectively block the actions of one receptor without affecting the other. Finally, it must be appreciated that activation of upstream effectors is very often associated with the amplification of downstream events so that activation of one signaling pathway often leads to activation of multiple downstream pathways. To date, few studies have attempted to examine systematically the sequence of activation for NGF. Thus, a number of studies are needed to further resolve the inconsistencies of the current literature and ascertain the mechanisms by which NGF produces peripheral sensitization.
- © American Society for Pharmacology and Experimental Theraputics 2007
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵

Michael R. Vasko, Ph.D., is Paul Stark Professor of Pharmacology and Departmental Chair of Pharmacology and Toxicology at Indiana University School of Medicine. Address correspondence to MRV. E-mail vaskom{at}iupui.edu; fax 317-274-1560.

Grant Nicol, Ph.D., is the Showalter Professor of Pharmacology and Toxicology at the Indiana University School of Medicine. His work focuses on the modulation of ion channel activity and excitability by inflammatory mediators in sensory neurons.

