A NEW LOOK AT THE XANTHINE ALKALOIDS
- Stanley Scheindlin, DSc
The xanthine alkaloids—caffeine, theobromine, and theophylline—are closely related compounds found in a variety of plants indigenous to several continents. They are not true alkaloids, as they are only weakly basic, and possess some acidic properties. Their structural formulas (Figure 1⇓) include a purine ring, as found in the adenine and guanine nucleotides of DNA. The placement of the N-methyl groups determines the specific pharmacological profile of each compound. The xanthines are not of great current importance as medicinals, but their history and involvement in the general culture makes them of continuing interest.
Discovery of Caffeine
There is an intriguing story behind the discovery of caffeine, connecting the renowned German genius Goethe with an obscure young chemist, Friedlieb Runge. Johann Wolfgang von Goethe (1749–1832)—poet, novelist, playwright, and savant—was actively interested in the natural sciences. He was also a capable and respected administrator. Beginning in 1775, he served as Privy Councillor and Minister of State to Duke Karl August, at Weimar, in the duchy of Sachsen-Weimar-Eisenach. His duties included overseeing educational institutions, including the University at Jena only twelve miles from Weimar (1). Goethe was instrumental in establishing a chemistry laboratory at Jena, visited it frequently, and desired to be briefed about the latest discoveries, such as stoichiometry and electromagnetism (1).
In 1819, Friedlieb Runge, age twenty-five, completed his medical course at Jena. His dissertation dealt with extracts of the Solanaceous plants deadly nightshade (belladonna), henbane (hyoscyamus), and thornapple (stramonium), and he discovered their mydriatic effect, using cats for his experiments. This in vivo test, involving felines, has been adopted in forensic medicine as a general test for atropine-like alkaloids (2). Runge’s analytical chemistry professor, Johann Wolfgang Dobereiner, was also Goethe’s advisor on chemistry. Dobereiner informed Goethe about Runge’s research and his remarkable ability to dilate the cat’s pupil. Excited, Goethe requested to see the experiment and an appointment was set up. Picture, then, young Runge striding purposefully across the campus, wearing a frock coat and top hat borrowed for the occasion, carrying a henbane extract in one hand, and clutching a protesting cat under his other arm. Undergraduates, to whom he is already a figure of fun, shouting “Hier kommt Doctor Gift” (here comes Doctor Poison). He stops and explains the reason for his formal garb. The students are awed and the joking ceases.

Goethe

Runge

Thornapple

Henbane

Deadly Nightshade

Dobereiner
Goethe was highly gratified by Runge’s pharmacologic demonstration and questioned him in detail about his researches. Seemingly as an afterthought, as Runge was leaving, Goethe handed him a packet of Arabian coffee beans and suggested that this “rather rare” vegetable product might yield interesting chemical results. Runge went to work on the beans, and before the year was over, discovered a crystalline substance, which was named Koffein (2). Runge reported his findings in his newly founded but short-lived journal (in English translation) Newest Phytochemical Discoveries. From this point on, Runge, who went on to make other important discoveries, was no longer involved with caffeine (2). It was left to Emil Fischer, who reported the structure of caffeine in 1882 and accomplished its synthesis in 1885 (3).
Caffeine in the Cultural Context
Next to alcohol, caffeine is the most universally valued mood-improving substance. Every human society produces consumable alcohol from any readily available source of fermentable sugar (or carbohydrate that can be hydrolyzed to a fermentable sugar). In contrast, caffeine exists preformed in various members of the vegetable kingdom (Table 1⇓) that are processed into palatable drinks.
Wine is consumed largely in a social context and, to some extent, in religious ceremonies. Similarly, social ceremonies have grown up around caffeinated drinks. For example, tea has given rise to the Japanese tea ceremony and to English high tea. The sipping of coffee, although less steeped in tradition in Western societies, is equally pervasive. Coffee is consumed in the work place at morning and afternoon “coffee breaks,” as well as at the chain coffee houses, which have proliferated in recent years. Although patrons do socialize at these coffee houses, many pursue their academic studies or do business via lap-top and cell phone while nursing a coffee. Even people who drink decaffeinated coffee or tea, or some form of herbal tea, are paying homage to traditions derived from caffeine. It is notable that the word “café” is used to designate any informal eatery, even one where alcohol is purveyed. The involvement of caffeine in cultures around the world is discussed in interesting detail in a book published a few years ago (4). [Interested readers are encouraged to read the review of “The World of Caffeine” in Molecular Interventions (5).]
Caffeine: Human Pharmacokinetics
When one drinks caffeine-containing beverages, caffeine is completely absorbed, reaching a peak blood level in thirty to sixty minutes and rapidly crossing the blood–brain barrier. Its average half-life is four to six hours and ranges from two to twelve hours, depending on the (patho)physiological state of the individual. For example, the half-life is shorter in smokers and longer in pregnancy or chronic liver disease. Caffeine is extensively metabolized, first by demethylation to one of three possible dimethyl xanthines, and further by acetylation, 8-hydroxylation, and oxidation. Neonates, from birth to three months, cannot demethylate caffeine; they eliminate it by renal excretion, with half-lives of up to 100 hours. A number of drug interactions are known. Thus, caffeine metabolism is inhibited by alcohol, cimetidine, disulfiram, mexiletine, norfloxacin, and oral contraceptives but is accelerated by tobacco smoke (6).
Coffea arabica
Caffeine: Pharmacology
Caffeine’s most prominent pharmacologic action is to stimulate the central nervous system (CNS), increasing arousal and vigilance, reducing fatigue, and decreasing motor reaction time for some tasks. It may interfere with sleep, primarily in light caffeine users, and it tends to decrease cerebral blood flow. Its cardiovascular (CV) effect is a 5–10 mm increase in blood pressure via peripheral vasoconstriction, a slight decrease in heart rate, and to cause the systemic release of epinephrine, norepinephrine, and renin. Caffeine increases the respiratory rate by sensitizing the medullary respiratory center to CO2. This is the basis for its respiratory stimulant effect in newborns with recurrent episodes of apnea (6). Additionally, caffeine can weakly dilate the lung bronchi. Gastric secretion of acid and pepsin is also stimulated by caffeine and may contribute to acid reflux symptoms by decreasing the lower esophageal sphincter pressure. Other effects include caffeine’s ability to increase metabolic rate and circulating concentrations of free fatty acids, cortisol, and blood glucose (6).
Tolerance to the effects of caffeine rapidly develops. The effect of caffeine in any individual depends on how much he or she usually consumes, the schedule of caffeine intake, and the person’s individual half-life for caffeine. When caffeine is discontinued, withdrawal symptoms occur, mainly including headache and fatigue, and less commonly anxiety, impaired psychomotor performance, nausea or vomiting, or an intense desire for coffee (6). Thus, it is well established that caffeine causes physical dependence; the extent to which it may be addictive is a matter of uncertainty.
Similarly, the possible association of caffeine or coffee consumption with various diseases has been extensively investigated. The conditions studied include CV disease factors such as adverse changes in blood lipids and arrhythmias; decreased fertility, low birth weight, and neonatal withdrawal symptoms; mild psychiatric disturbances such as nervousness or insomnia; aggravation of existing agoraphobia, panic disorders, or psychoses (6). To date none of these studies has produced the proverbial “smoking gun;” thus, caffeine consumption continues unabated.
Caffeine: Therapeutic Uses
Caffeine has long been used as an adjuvant in analgesic products, where it is believed to enhance gastrointestinal absorption of the analgesics and potentiate their activity. It was a component of an analgesic-antipyretic tablet containing aspirin (227 mg), phenacetin (162 mg), and caffeine (32 mg) first marketed by Burroughs Wellcome as Empirin Compound®. The combination was widely copied generically as APC. The seemingly odd milligram-composition arises from its having been originally formulated by the apothecary system of weights. Because 64.8 mg equal 1 grain, the composition translates to 3.5 grains of aspirin, 2.5 grains of phenacetin, and 0.5 grains of caffeine.
In the 1960s, long-term use of APC was linked with serious renal pathologies (e.g., interstitial nephritis, pyelonephritis, and medullary necrosis). Phenacetin was deemed to be the culprit, although much medical opinion held that chronic use and misuse of the total product was to blame. The FDA first required that OTC phenacetin products carry the following label warning: “This medication may damage the kidneys when used in large amounts or for a long period of time...” (7). Ultimately, the FDA banned phenacetin entirely; but by the time this happened, manufacturers had already eliminated phenacetin from their products in anticipation of the ban (8).
Following the demise of APC, “AAC” products, in which phenacetin was replaced by acetaminophen (APAP), were marketed but these never achieved the popularity of APC. Nevertheless, one AAC product deserves special mention. Excedrin® Migraine [containing aspirin (250 mg), APAP (250 mg), and caffeine (65 mg)] was approved in 1998 under a New Drug Application (NDA). For a combination-product NDA to be approved, each of the active ingredients must be shown to contribute to the product’s effectiveness. At least one generic AAC has also been approved. In clinical trials, AAC provided significant benefits of pain relief and reduction of other migraine-associated symptoms in patients with mild-to-moderate migraine attacks (9).
Generally, over-the-counter (OTC) products are not marketed under NDAs but under OTC “monographs” established by the FDA. The monograph covering OTC Internal Analgesics is not yet final, but it allows for 65 mg of caffeine per dosage unit to be added to analgesics such as aspirin and APAP. Another OTC indication for caffeine is recognized in the monograph “Stimulant Drug Products for Over-the-Counter Human Use.” The only stimulant approved to “help restore mental alertness or wakefulness when experiencing fatigue or drowsiness” is caffeine, at a dosage of 100–200 mg (10).
A prescription-only use of caffeine is the treatment of apnea in newborns. In premature babies, especially those born after twenty-eight to less than thirty-three weeks of gestation, episodes of apnea tend to occur frequently. Both caffeine and theophylline are comparably effective in regularizing the breathing, but caffeine is easier to administer, has a wider therapeutic index, and produces fewer peripheral effects (6, 11). For this purpose, it is administered intravenously (iv) or by mouth, in a combination with citric acid, which solubilizes the caffeine (12).
Theobromine
Sneader’s excellent history of drug discovery erroneously states that theobromine was isolated in 1878 (3). Actually, it was in January 1842 that M.A. Woskresensky reported the isolation of theobromine from a hot water extract of cacao seeds (13). Theobromine is the predominant alkaloid of the chocolate bean, but unlike caffeine, has virtually no central stimulant action. Nonetheless, it has been suggested that theobromine is habit-forming, which may explain why people become “chocoholics” (14). In an interesting quirk of nature, dogs metabolize theobromine so slowly that they may be poisoned by eating chocolate (14).
Theobromine: Actions and Uses
Theobromine’s main pharmacologic action, as a diuretic, was discovered in the 1880s at the University of Strassburg (15). Subsequently, it came into clinical use as a diuretic, myocardial stimulant, and coronary and peripheral vasodilator (16). To increase the solubility of theobromine and minimize gastric irritation, double salts were prepared by mixing it with equimolar amounts of sodium acetate, sodium salicylate, or calcium salicylate—although one would think that salicylates would add to gastric discomfort. Since the introduction of more effective diuretics (e.g., thiazides), theobromine has fallen out of use and is no longer recognized in the United States Pharmacopeia (USP).
Nevertheless, theobromine continues to be a subject of research. A recent study deals with its possible antitussive use. Cough is a troublesome and widespread symptom, for which remarkably few safe and effective remedies are available. Usmani et al. noticed that, in a series of substituted xanthine compounds prepared as possible antiasthmatics, compounds with no substituent on the N1 atom showed an antitussive effect. They then began to examine theobromine, the only one of the xanthine alkaloids in which N1 is unsubstituted. The authors first investigated the effect of theobromine, as compared to codeine and placebo, on citric acid-induced cough in guinea pigs, finding a dose-dependent antitussive effect: at a dose of 32 mg/kg the theobromine effect was statistically significant for up to four hours. Next, they found that theobromine inhibits capsaicin-induced cough in ten healthy volunteers. To look into the mechanism of action, they studied perfused, isolated preparations of guinea pig vagus and human vagus nerves. In both types of preparations, theobromine inhibited capsaicin-induced nerve depolarization in a concentration-dependent manner. They concluded that the cough reflex was being inhibited via a peripherally mediated action (17). If the suggestive work of Usmani’s group is followed up, theobromine or one of its derivatives may become an important antitussive agent in the coming years.
Theophylline: Historical Overview

Minkowski
Theophylline, first isolated in 1888 from a tea extract (18) (Box 1), was discovered much later than the other xanthines, presumably because its presence was masked by the relatively large amount of caffeine contained in tea. Clinical interest developed as soon as sufficient quantities became available through synthesis. By 1907, Minkowski and other German doctors had brought it into use as a diuretic [in cases of edema of both cardiac and renal origins (19)], and by 1918, it was included in the USP (20).
Methylxanthine Extraction
It is notable that tea and coffee, the two major caffeinated beverages, are made by extraction processes used in pharmacy. Tea leaves are extracted by steeping them in boiling water, with no further heating, a process called infusion. Ground coffee beans are packed onto a filter and extracted by running boiling water through them, a process called percolation.
Unlike most of our modern drugs, theophylline has been used medicinally for a full century, and has had a varied career. In the beginning, its diuretic action was at the fore. Then, as its cardiac stimulant and bronchodilator actions were recognized, it came into use for paroxysmal nocturnal dyspnea resulting from left ventricular failure, Cheyne-Stokes respiration, status asthmaticus, and prophylaxis of asthma attacks (21).
In the 1970s and 1980s, as the science of pharmacokinetics (PK) developed, theophylline’s PK was extensively studied, and the plasma levels associated with bronchodilator effect and toxicity, respectively, were elucidated. Its medicinal use was placed on a rational footing, and it became the drug of choice for the management of asthma. Nearly concurrently, new classes of drugs were being developed that changed the face of asthma therapy and have largely eclipsed theophylline.
Theophylline: Pharmacology, PK, and Derivatives
Although theophylline shares some of the stimulant properties of caffeine and the diuretic effects of theobromine, its bronchodilator action has been of greatest importance in modern medicine. Theophylline relaxes airway smooth muscle, thus exerting a therapeutic effect in asthma. The mechanism of action is based on a non-selective inhibition of cyclic nucleotide phosphodiesterases (PDEs), preventing breakdown of cyclic AMP and cyclic GMP. Another important action is theophylline’s competitive antagonism at adenosine receptors—adenosine is known to cause bronchoconstriction in asthmatics. Theophylline also inhibits synthesis and secretion of inflammatory mediators from various types of cells; it has been proposed, but not yet proven, that this anti-inflammatory effect may be relevant to the drug’s therapeutic action (22).
From liquids or uncoated tablets, theophylline is rapidly and completely absorbed, producing peak plasma levels within two hours. Administration with food slows the rate but does not limit the extent of absorption. Sleep, or even lying down, may significantly reduce the rate and extent of absorption. At therapeutic concentrations theophylline is about 60% protein-bound. It is metabolized in the liver, only about 15% being excreted unchanged. The rate of elimination varies widely among individuals, partially owing to genetic factors. The half-life is typically eight to nine hours in adults, and about 3.5 hours in young children (22). The therapeutic range for theophylline’s bronchodilator action has been established at 10–20 mg/L. Above 20mg/L, toxic effects may occur, including tachycardia, severe restlessness, agitation, emesis, and occasionally seizures (23). Desired plasma concentrations are obtained by careful dose adjustment and periodic blood testing.
From the beginning, theophylline’s limited water-solubility (1 g in 120 mL) was recognized as a problem, especially as it needed to be administered by injection in life-threatening conditions. Accordingly, more soluble complexes or salts were introduced, which dissociated in water to yield free theophylline. The most notable of the complexes is theophylline ethylenediamine (aminophylline), which is almost as old (1908) as theophylline itself. An example of a true salt is choline theophyllinate (oxtryphylline). Highly soluble covalent derivatives were also synthesized [e.g., 7-dihydroxypropyl theophylline (dyphylline, Neothylline®)], introduced in the 1950s. These compounds exhibit their own PK and pharmacologic characteristics. Dyphylline, for example, has a half-life of only 2.5 hours and a lower potency than theophylline; thus, it never attained a strong position in therapeutics.
Non-Bronchodilator Derivatives
Two methylxanthine derivatives have found a place in medicine for conditions unrelated to the indications for theophylline: 8-chlorotheophylline and 1-(5-oxohexyl) theobromine (pentoxifylline). 8-Chlorotheophylline has always been used in combination with the oldest anti-histamine [diphenhydramine (Benadryl®)] in the popular antinauseant–antiemetic Dramamine® (dimenhydrinate). This product is described as a salt, diphenhydramine 8-chlorotheophyllinate.
Benadryl®, like the other H1 receptor histamine antagonists, has drowsiness as a major side effect. In fact, a number of OTC products employ it, alone or in combination with an analgesic, as a sleep-aid. Generally, however, this side effect is troublesome, and the H2 receptor antagonists have been developed to obviate it. In the 1940s, the GD Searle company combined diphenhydramine with 8-chlorotheophylline, a mild stimulant. Although this did not solve the drowsiness problem, it turned out to have a desirable action as an anti-motion-sickness agent (24).
Testing the product for antihistaminic action, Gay and Carliner at Johns Hopkins made a serendipitous discovery. They administered Dramamine® to a patient plagued by hives. She reported that, for the first time in years, she rode a trolley car without feeling sick (25). In subsequent trials, the drug was found effective against other forms of motion sickness and against the nausea of pregnancy.
Motion sickness has been a problem from antiquity. The Bible does not record anyone becoming queasy while riding on a camel, but seasickness must have occurred since men first sailed the oceans. In the 20th century new forms of motion sickness—rail sickness (trains and trolleys), car sickness (motor vehicles), and air sickness (planes)—were added to seasickness. A review of the subject by Tyler and Bard (26) indicates that prior to World War II little good research had been done on the subject. The only solid discovery was that the vestibular apparatus of the ear is essential in producing motion sickness. Early in the war, the military realized that moving large numbers of men by sea and air would produce high rates of motion sickness, and more definitive experimental work was initiated.
The effectiveness of Dramamine® was tested in a placebo controlled trial carried out on a troopship sailing from New York to Bremerhaven, Germany in November 1947. In a rough ten-day crossing, 4% of the soldiers given Dramamine® became sick, compared with 25% of those on placebo (24). Interestingly, Tyler and Bard (26) point out the design flaws of the troopship study, much as the highly sophisticated designs of current clinical trials are picked apart by commentators in the medical journals. Nevertheless, Dramamine® is deemed an effective product, and 8-chlorotheophylline has been a key ingredient of a viable medicinal for six decades—no mean feat in this era of rapid drug obsolescence.
Pentoxifylline (Trental®)
Pentoxifylline, a theobromine molecule substituted on N1 with an aliphatic ketone moiety, is inactive as a CNS stimulant or bronchodilator. It was introduced as a blood viscosity-reducing agent in peripheral vascular disease. Specifically, it is indicated for symptomatic relief of intermittent claudication, an arterial spasm causing painful cramping of the legs, and lameness. In clinical trials, it lengthened the distance patients could walk before the onset of claudication, and enhanced blood flow in their ischemic limbs. It appears, however, that only 20 to 30% of patients on this drug experience long-lasting benefit (27).
It was surprising to find pentoxifylline as the subject of a recent dermatologic clinical trial—a randomized, double-blind, placebo-controlled trial in the treatment of recurrent aphthous stomatitis (canker sores of the oral mucosa). The authors concluded that the drug showed limited benefit, and could not be recommended as firstline treatment (28). A second strike of mediocrity!
Theophylline: The Present and the Future
A number of new classes of drugs, developed in the last thirty years, have reshaped asthma therapy. Short-acting β2-adrenergic receptor agonists (e.g., albuterol) are used in acute attacks to reverse bronchospasm, inhaled glucocorticoids (beclomethasone, budesonide) are given to reduce frequency and severity of attacks; and newer anticholinergic agents (e.g., ipratropium) and leukotriene-receptor antagonists (e.g., montelukast) have proved to be helpful adjuncts. As a result, international guidelines have relegated theophylline to third-line therapy in both asthma and chronic obstructive pulmonary disease (COPD). This is the case in developed countries, where the cost of the new drugs is no obstacle, but inexpensive theophylline continues to be widely prescribed in other parts of the world.
Barnes (23) has reviewed the ongoing research on theophylline from the 1980s to the early years of the current decade. Much of the research has delved into theophylline’s molecular mechanisms of action. Nonselective inhibition of the PDE enzymes seems to be the main mechanism for the drug’s bronchodilator action, but may also be responsible for common side effects nausea and headache. Theophylline is also a potent inhibitor of adenosine receptors. It has not been proven that this helps the anti-asthma effect, but it may account for such serious side-effects as seizures and cardiac arrhythmias (23).
Of great interest are the emerging findings regarding theophylline’s anti-inflammatory effects. One of the main mechanisms of this action is the activation by theophylline of histone deacetylase (HDAC) activity, which suppresses the expression of inflammatory genes. Whereas theophylline alone has weak anti-inflammatory action, in low concentrations it potentiates the effects of corticosteroids. It may thus combat the resistance to corticosteroids seen in patients with COPD or severe asthma. Potentiation of the corticosteroid effect has been shown in vitro, and low-dose theophylline added to inhaled corticosteroid has shown benefit in clinical studies of patients with asthma. Barnes suggests that low doses of theophylline, producing plasma levels of 5–10 mg/L (half the concentration used for bronchodilation), will potentiate the action of inhaled corticoids, especially in patients with severe asthma or COPD. At these low plasma concentrations, side effects and drug interactions are largely avoided (23). Further clinical trials are needed to establish the viability of this approach, which would require a rethinking of the way theophylline and corticoids are used in the treatment of airway disease.
Research on new theophylline derivatives is also being carried out, particularly in Europe. One such derivative, doxofylline, has a dioxolane substituent on N7 of theophylline. Doxofylline is an active PDE inhibitor, but has much lower affinity for adenosine receptors. Thus, it retains a bronchodilator effect but does not share theophylline’s effects on cardiac rhythm. This might be favorable for patients with chronic respiratory disease and pre-existing arrhythmias or in those with airflow obstruction secondary to cardiac disease (29).
The future thus may bring a resurgence of theophylline, or the introduction of a new structural modification as an improvement on the parent drug.
Botanical Sources of Caffeine
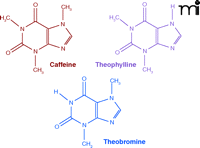
The chemical structures of three common methylxanthines: caffeine, theophylline, and theobromine.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Stanley Scheindlin, DSc, holds a BS in pharmacy from Temple University and graduate degrees in pharmaceutical chemistry from Philadelphia College of Pharmacy and Science (now University of Sciences in Philadelphia). His academic research dealt with plant constituents and chemical interactions of vitamins. In his pharmaceutical industry career, he handled new drug formulation development, and later regulatory affairs, presiding over the filing of about 100 generic new drug applications and two innovative drug applications. Now retired, his activities include volunteer work, consulting, and writing Reflections pieces for this journal.

