Bound to Be Different: Neurotransmitter Transporters Meet Their Bacterial Cousins
- L. Keith Henry1,4,5,
- Jens Meiler1,2,5 and
- Randy D. Blakely1,3,4
The neurotransmitter transporters belonging to the solute carrier 6 (SLC6) family, including the γ-aminobutyric acid (GAT), norepinephrine (NET), serotonin (SERT) and dopamine (DAT) transporters comprise some of the most clinically relevant drug targets today. These Na+, Cl−–dependent transporters primarily function following neurotransmission to reset neuronal signaling by transporting neurotransmitter out of the synapse and back into the pre-synaptic neuron. Other members of the family include amino-acid transporters that span the evolutionary spectrum from archaebacteria to man. In four recent and thought-provoking studies of human SLC6 transporters and their somewhat similar bacterial analogs, two very important aspects of transporter physiology have been explored. First, studies from the labs of Honig (1) and Kanner (2) have tracked down an elusive binding site for Cl− that facilitates neurotransmitter transport using structural differences evident with bacterial family members (e.g., the Aquifex aeolicus leucine transporter LeuTAa) that lack Cl− dependence. Second, the laboratories of Gouaux (3) and Wang (4) have solved crystal structures of antidepressant-bound LeuTAa, revealing a surprising mode of drug interaction that may have relevance for medication development. These four studies have exploited the high-resolution crystal structure for LeuTAa (5), whereby prior biochemical, pharmacological, and electrophysiological data can be mapped onto a rational 3-D scaffold to illuminate ill-defined aspects of transporter structure-function. Whereas overall, LeuTAa shares approximately ∼20% amino acid sequence identity with mammalian neurotransmitter transporters, much higher identity is evident near the substrate binding site (∼45%), encouraging the generation of homology-based models for eukaryotic homologs.
The groundbreaking structural studies of LeuTAa by Yamashita et al. (5) revealed its arrangement of transmembrane (TM) helices, a binding site for leucine midway through the 12-TM-spanning structure, residues responsible for Na+ coordination, and putative outer and inner gates that limit substrate flux. Although this is a substantial accomplishment, we await structures of eukaryotic family members to validate and extend the generality of these findings. In the meantime, the structure of LeuTAa is quite instructive regarding how sequence divergence has evolved to establish the distinct properties of neurotransmitter transporters. The study of sequence divergence between LeuTAa and human SLC6 family transporters should soon inform us as to how and why neurotransmitter transporters evolved a reliance on extracellular Cl− to propel the transport cycle; what residue changes and helical rearrangements give rise to recognition of different substrates; and how drugs such as antidepressants, cocaine, and amphetamines halt (or reverse) the transport process.
One of the hallmarks of the SLC6 transporters is their dependence on Na+ and Cl− for function (6). Because Na+ gradients are thought to energize the transport cycle, this ion’s interaction with SLC6 family members has received the lion’s share of attention. Indeed, some studies point to the idea that Cl− is more a regulator of transport rather than being coupled energetically to substrate movement (7). Regardless, Cl− binding appears essential for maximal activity of many SLC6 transporters. Notably, the LeuTAa structure reveals multiple Na+ binding sites but lacks a Cl− binding site that can interact with Na+ or the substrate, in keeping with the LeuTAa’s Cl−-independent leucine transport activity. Two independent groups, working on the assumption that LeuTAa is a good structural model for eukaryotic members of the SLC6 family, scanned the LeuTAa structure for residues that might have diverged to permit the formation of a Cl− binding site. Forrest et al. (1), performed pKA analysis, homology modeling, and site-directed mutagenesis to implicate the presence of a native negative charge in LeuTAa and Aedes aegypti phenylalanine transporter (AaAAT1), proximal to the core of the protein, lacking in the sequences of eukaryotic SERT and GAT1––transporters that presumably acquire this negative charge through Cl− binding. Zomot et al. (2) undertook similar methods to identify putative Cl− binding sites in GAT1 and GAT4 GABA transporters , and DAT, but relied upon an understanding of how Cl− coordination is established in chloride channels (Cl-C) (8). Utilizing these simple, yet elegant, approaches, the two groups converged on common structural elements necessary to achieve Cl− dependence in eukaryotic SLC6 family members. Besides a substitution of a charged residue [e.g., E290 in LeuTAa becomes S372 in hSERT and S331 in GAT1 (see Figure 1⇓)], eukaryotic transporters faciliate Cl− binding through Tyr and Ser residues that replicate aspects of the anion-binding sites of Cl-C channels and halorhodopsin. An intriguing characteristic shared by the LeuTAa structure and the Cl-C channels is the presence of two helices with unwound regions midway through the membrane that exposes both (+) and (−) dipoles around the substrate binding site. Perhaps this common feature of Cl− channels and SLC6 transporters plays a role not only in substrate accommodation but also in propagation of conformational changes that ensue following substrate/ion binding. These studies lay a framework on which previous data from SLC6 ion-dependence studies can be re-evaluated and expanded for greater understanding of the process of ion coupling and its physiological role in support of neuronal signaling. Presuming, based on the data discussed above, that we know with reasonable certainty where Cl− is located in the transporter, we might be able to clarify why Cl− is needed for neurotransmitter transport in the first place. Some researchers suggest that neurotransmitter transporter–dependent Cl− fluxes may function to regulate neuronal activity (9, 10). Although many observations of charge flux through neurotransmitter transporters exist, it remains unclear whether Cl− actually moves through the transporter or, alternatively, participates in local network coordinations required to gate neurotransmitter flux (or both). Our own results (11) indicate that in hSERT, mutations near the proposed Cl− and 5-HT binding sites can eliminate Cl− dependence with little or no impact on 5-HT transport. We must also recognize that ion-flux can be greatly perturbed by alterations in transporter-associated proteins (12), suggesting that the answer may be context dependent and relate to the evolution of transporter regulatory pathways in eukaryotes. These distinctions remind us of the differences between “ion dependence” and “ion coupling,” differences that ultimately must be resolved before we truly understand how neurotransmitter transporter molecules work and are regulated.
Another set of high-profile studies involves new crystal structures of the LeuTAa protein bound to antidepressant molecules. Of course, we would prefer to have structures of neurotransmitter transporters bound to these antagonists, but these transporters have not yielded to atomic-level structural analysis. One set of structures (3) comes from the lab that originally introduced us to the LeuTAa structure and reveals the binding of this transporter to the tricyclic antidepressants (TCAs) clomipramine (CMI), desipramine (DMI) and imipramine (IMI). Similarly, Zhou et al. (4) report the binding of DMI to LeuTAa. A general characteristic of TCA binding revealed by these studies is that binding results in only small changes in the overall structure of the transporter when compared to structural changes in leucine-bound LeuTAa structure [root-mean-square deviation (RMSD) of 0.2 Å for all non-hydrogen atoms]. Another common finding between these two studies reveals that TCAs bind to the extracellular portion of the proposed permeation pathway just above the upper gate formed by R30 and D404 (Figure 2⇓). This mode of binding is quite striking as it neither protrudes into nor disturbs the substrate-binding pocket. In the LeuTAa leucine-bound structure, R30 and D404 interact via an H-bond network formed by two water molecules. This interaction is altered in the TCA-bound structures. Here the binding of the TCAs excludes the two water molecules, promoting a stabilizing salt-bridge interaction between R30 and D404 that appears to preclude movement of leucine through the outer aspects of the permeation pathway. Indeed, kinetic experiments by Singh et al. (3) demonstrate that TCA binding is noncompetitive and greatly reduces the dissociation rate of prebound substrate. It is noteworthy that the CMI, DMI, and IMI co-crystals solved by Singh et al. (3) were at significantly higher resolutions (1.7–1.9 Å) as compared to the structure (2.9 Å) solved by Zhou et al. (4). The increased resolution of the Singh et al. structures allow for greater confidence in interpreting the presence of small movements as compared to the original LeuTAa structure, although many of the observed movements were seen by both research groups.
How, then, does the TCA binding site in LeuTAa relate to the binding site for these drugs and other antidepressants in neurotransmitter transporters? This is especially relevant given the low potency of TCAs for leucine transport inhibition (CMI=250 μM, IMI=2 mM) in LeuTAa vs their much higher potency in antagonism of neurotransmitter transport (CMI=1.7 nM, IMI=7.7 nM, hSERT). Additionally, as Singh et al. demonstrate, TCA binding to LeuTAa exhibits noncompetitive kinetics whereas these and other antagonists display competitive inhibition kinetics against SERT and NET (13, 14). Regarding this question, the two studies take decidedly different positions. Zhou et al. claim that their LeuT-DMI structure reveals a conserved binding site and inhibition mechanism, whereas Singh et al. link their results to a more general explanation for how noncompetitive inhibition can arise from allosteric mechanisms. Zhou et al. support their claims through site-directed mutagenesis studies, where a gain of potency (2- to 5-fold) for DMI is generated in hSERT and hDAT after conversion of TCA-binding residues (as defined in LeuTAa, DMI=80μM) to those identified in hNET, a high-affinity DMI target (DMI=4 nM). The position of the TCA-binding site in LeuTAa places it in a less conserved area among the SLC6 transporters, as compared to the more highly conserved substrate binding site. From this perspective, an important experiment missing from the Zhou et al. study is evaluation of reciprocal mutation of the residues in LeuTAa that were important, based on the crystal structure. Arguing against the generalization of these observations to neurotransmitter transporters, conversion of two residues in human and mouse SERT within the proposed binding site for 5-HT (Y95, I172) to their Drosophila SERT identities (F90 and M167 respectively) reduces antidepressant potency for 5-HT uptake inhibition by as much as 10- and 1000-fold in the single Y95F and I172M mutants, respectively (15). Furthermore, an hSERT double mutant containing both Y95F and I172M substitutions gives a 10,000-fold loss in potency for the antidepressant citalopram. Reciprocal mutations in Drosophila SERT (i.e., F90 and M167 to Y95 and I172, respectively) yield a gain of potency for antidepressants, these data argue that high-affinity recognition of SERT-selective reuptake inhibitors (SSRIs) occurs through residues lining the 5-HT-binding pocket. For TCAs, a comparable argument exists because CMI potency in the I172M mutant is reduced by 178-fold (15).
Can these two binding modes for SLC6 inhibitors, one above the closed outer gate and one overlapping with the substrate binding site, be reconciled? Even with a 10,000-fold loss of potency for citalopram in the hSERT Y95F/I172M mutant, measurable inhibition of 5-HT transport by citalopram remains. This residual, low-affinity inhibition could be explained by the presence of a second citalopram binding site. Future studies should explore whether the remaining citalopram potency observed in the Y95F/I172M mutant is due to the residues analogous to the TCA binding determinants in LeuTAa. Perhaps a more interesting finding is the total lack of sensitivity of the SSRI paroxetine and the TCA tianeptine to the I172M substitution (15). Although slightly different conformations may support high-affinity binding of these drugs at the substrate binding site, an alternative explanation is that they interact at more distal sites. Such an explanation may also relate to findings that the R isomer of citalopram can impact the dissociation rate of the S isomer from its high affinity site (16), possibly explaining suggestions of surprisingly superior antidepressant efficacy of S-citalopram over the racemic mixture (17). For example, R-citalopram could stabilize an open outer gate in hSERT while also preventing conformational changes required for 5-HT transport. With the limited sequence identity evident between bacterial and eukaryotic neurotransmitter transporters, distinctions in transporter binding sites were bound to be evident. On the one hand, variations in structure have led to new ion binding sites, whose functional role can now be directly assessed. On the other hand, unexpected and possibly conserved modes of transport inhibition have emerged. What the field really needs are X-ray structures of eukaryotic neurotransmitter transporters in the absence and presence of Na+, Cl−, neurotransmitters and antagonists. As such structures are solved, they are bound to reveal surprises regarding the inner workings of neurotransmitter transporters predicted here from bacterial cousins. We eagerly await their arrival.
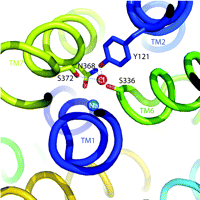
Cl− binding site in hSERT. Illustration depicts hSERT labeled helices (TM) and residue side chains identified by Forrest et al. (1) to coordinate Cl− binding in the human serotonin transporter (hSERT) (stick rendering). The Cl− (red) and Na+ (blue) ions are shown as labeled spheres. The figure was generated in Pymol (DeLano Scientific LLC, San Carlos,. California) using coordinates from a LeuTAa-hSERT homology model we have developed in our studies.
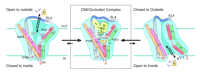
Schematic illustrating a mechanism for noncompetitive inhibition of LeuT by the TCAs. The cartoon labeled CMI/Occluded Complex depicts the coordination sites for clomipramine and substrate positioned in non-overlapping binding modes above and below the upper gate (salt bridge), respectively. See text for details. Adapted by permission from Macmillan Publishers Ltd: Nature448, 952–956, © 2007.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Jens Meiler, PhD, is an Assistant Professor of Chemistry, Pharmacology, and Biomedical Informatics at Vanderbilt University. He received his PhD in Chemistry from Frankfurt University, Germany, working with Christian Griesinger. He then trained with David Baker in the Department of Biochemistry at University of Washington, Seattle. Dr. Meiler’s laboratory in Nashville focuses on method development and application in computational structural biology. E-mail jens.meiler{at}vanderbilt.edu

L. Keith Henry, PhD, is currently a research instructor in the Department of Pharmacology at Vanderbilt University School of Medicine. He has recently accepted an assistant professor position at The University of North Dakota School of Medicine and Health Sciences in the Department of Pharmacology, Physiology and Therapeutics. Please address correspondence regarding this article to LKH. E-mail keith.henry{at}vanderbilt.edu phone 615 322–0122 fax 615 936–3040. Contact information after April 2008: E-mail khenry{at}medicine.nodak.edu; fax 701 777–4490.

Randy D. Blakely, PhD, is the Allan D. Bass Professor in the Department of Pharmacology and Director of the Center for Molecular Neuroscience at Vanderbilt University. E-mail randy.blakely{at}vanderbilt.edu

