The See-saw of Differentiation: Tipping the Chromatin Balance
Every cell in the body, with a few exceptions such as lymphocytes, carries exactly the same amount and type of genetic information. But despite their identical genome, cells are remarkably different, comparing, for example, their ability to form a one-meter long axon or to contract 140 times per minute. These phenotypic differences are determined by the way genetic information can be retrieved, which depends largely on the packaging of DNA into chromatin. During development, traversing from the pluripotent embryonal phase to the specialized somatic tissue stage, cells sort and package the genome into areas that are either highly accessible for transcription factors or into zones that are less open or even become permanently inaccessible. Thus, diverse chromatin states establish accessibility and prepare cells to respond differently to signals, resulting in tissue-specific expression patterns of either coding or non-coding transcripts of RNA.
What are the characteristics of the chromatin structure that make up the specific memory of a cell? Can we identify key chromatin patterns that alter the accessibility of genes during differentiation? The answer to this question is not only of profound significance for understanding developmental cell biology, but it also bears the hope that one day we may be able to specifically modulate accessibility in the genome to control cellular differentiation and to use it for therapeutic purposes.
Recent studies by Bernstein and Lander (1, 2) give now a first glimpse of what type of chromatin modifications may define the pluripotent versus the differentiated cellular state. They identified a chromatin pattern, termed “bivalent,” that appears to be unique for pluripotent cells and that resolves upon differentiation, consisting of the simultaneous presence of activation and repression chromatin marks.
Numerous epigenetic mechanisms have been identified that determine chromatin structure [for review, see (3–5)], including DNA methylation [the covalent linkage of a methyl group to cytosine in the context of a cytosine-phosphate-guanine (CpG) dinucleotide in mammals] and SNF1 chromatin remodeling complexes that may shift or displace nucleosomes (the smallest subunit of chromatin), or may loosen the interaction of DNA to histones (the core proteins of nucleosomes). In addition, many posttranslational modifications of the core histones (H2A, H2B, H3, and H4) have been identified that include acetylation, methylation, and phosphorylation. In particular, methylation of lysine (K) residues on H3 has gained attention for a variety of reasons: 1) methylation occurs at distinct lysine residues of H3 (such as H3K4 versus H3K9 or H3K27) and leads to distinct biologic consequences; 2) the degree of methylation at a specific lysine (mono-, di-, or tri- methylation such as H3K4me1,2,3) is associated with distinct functions; and 3) there exist unique histone methylases as well as demethylases (making the methylation process highly specific as well as reversible).
Though the complexity of histone modifications is astounding, large-scale whole-genome analysis has revealed certain global patterns. For example, to identify functional elements of the human genome, the Consortium of the ENCODE project (The Encyclopedia of DNA Elements) has examined about thirty megabasepairs (approximately 1%) of the human genome (6). With respect to chromatin structure the data showed a close association of DNaseI sensitivity, chromatin accessibility, and high amounts of H3K4me2 or H3K4me3 modifications. Thus, H3K4me3 can be predictive for active transcriptional start sites. On the other hand, analysis of higher-order chromatin domains showed that regions enriched for H3K27me3 were associated with transcriptionally repressed chromatin. Furthermore, recent evidence suggests that the polycomb repressive complex in Drosphila as well as in mammals, long known to mediate silencing of developmental genes, contains an H3K27 methyltransferase. On the other hand, the tri-thorax protein–containing complex that is required for activation of the same genes silenced by polycomb proteins is associated with an H3K4 methyltransferase (again,in Drosophila and in mammals) [see (7)]. Whereas H3K4 methylation can support transcription for example by recruiting SNF nucleosome remodeling complexes, H3K27 promotes chromatin compaction (8–10). In addition, interactions between the polycomb H3K27 methytransferase and DNA methylation have been reported suggesting a close intertwining of the two epigenetic silencing pathways (11–14). Thus, several studies suggest that H3K4me3 is closely linked with transcription whereas H3K27me3 is associated with repression. Both modifications appear (with a few exceptional loci) mutually exclusive. However, these studies were performed in differentiated somatic cells, in which genes appear to be either committed to an on or an off state.
Bernstein and Lander have investigated whether these marks differ in pluripotent cells such as embryonic stem (ES) cells. Originally, they examined regions rich in highly conserved non-coding elements (HCNE) in murine ES cells (1). These regions were homologous between human and mouse and enriched for genes encoding developmentally important transcription factors. They observed in the novel bivalent chromatin large domains of H3K27me3 (a repressive mark) harboring small regions of H3K4me3 (an active mark).
In a subsequent study, they extended their analysis to about 17,000 promoter regions and identified this bivalent mark at about 15% of the examined sites in pluripotent murine embryonal stem (ES) cells (2). By comparing ES cells to differentiated cell types (such as neuronal progenitor cells and embryonal fibroblasts), they found that the majority of these bivalent domains seemed to resolve upon differentiation. Although the presence of the bivalent domains in ES cells was associated with very little or no transcription, differentiated cells frequently showed either H3K4me3 linked to high transcription or H3K27 me3 (or no mark) together with gene silencing. Many of the bivalent domains were located at genes encoding transcription factors that play an important role in lineage specification during embryonic development. Thus, their data suggests a model in which bivalence functions to repress transcription but also to keep genes poised for future transcriptional activation when a developmental differentiation signal would induce a specific pathway. Depending on the lineage, this would result in either activation or suppression of the gene. Accordingly, the bivalent domain would mostly resolve into either H3K4me3 or H3K27me3 marks.
Based on Bernstein and Lander’s findings, one may expect that histone demethylases play a crucial role in differentiation because removal of either K4 or K27 methylation is an important step in resolving the bivalent state. Indeed, recent studies have demonstrated a crucial role for H3K27 demethylases during development (15, 16). Secondly, although more chromatin modifications are likely to be involved in maintaining the pluripotent state of stem cells, the model of bivalence is of remarkable simplicity and it supports a scheme of reversibility (Figure 1⇓).
Since the cloning of Dolly, researchers have known that differentiated somatic cells can, in principle, be reprogrammed to a pluripotent stage, though it involved unknown factors present in oocytes. In seminal work by Yamanaka’s group, some of the factors that induce pluripotency have now been identified (17). Using murine skin fibroblasts, his group demonstrated that the expression of only four transcription factors––Sox2, c-Myc, Oct4, and Klf4––can induce the manufacture of pluripotent stem (iPS) cells. These transcription factors were known to be important for maintaining the pluripotency and self-renewal of stem cells, but it had not been previously demonstrated that the factors could invoke pluripotency from differentiated cells. The iPS cells showed similar gene expression to ES cells, and ultimately it was demonstrated that iPS cells are pluripotent because viable chimeric mice could be generated and germ-line transmission in mice was achieved (18, 19).
Meanwhile, subsequent studies demonstrated that not only murine cells but also human cell cultures can be induced to become stem cells (20–22). Furthermore, the presence of the four factors is not obligatory; because c-Myc and Klf4 are dispensable in skin fibroblasts, other gene combinations may be effective, too. And besides skin fibroblasts, other cell types (such as synoviocytes) can be used (20, 23).
What is the molecular mechanism by which pluripotent cells are induced from skin fibroblasts? And how does this relate to the bivalent chromatin pattern found in pluripotent ES cells? A limited analysis on only a few selected genes suggests that reprogramming of fibroblasts, indeed, involves the establishment of a pluri-potent chromatin pattern. For example, a number of fibroblast specific genes that showed only H3K4me3 also gained H3K27me3 upon “reprogramming” into iPS cells. In contrast, genes that were usually silenced in fibroblasts gained a H3K4me3 mark during de-differentiation, such that the selected sites presented the bivalent pattern characteristic for pluripotency (19, 20). In addition, the DNA methylation profile at the murine Oct4 and the Nanog promoter regions was extinguished during reprogramming and resembled that of pluripotent cells. Analysis of the sequences that were present at the bivalent domains revealed that more than 50% of the domains harbor at least one of the binding sites for the transcription factors Oct4, Nanog, and Sox2. This raises the possiblity that binding of these transcription factors may contribute in maintaining these sites in a poised bivalent state.
The possibility of generatinge patient-tailored iPS cells will undoubtedly spur research using in vitro differentiation models, accelerate our understanding of developmental biology, and circumvent some of the ethical considerations involved in the use of human oocytes or embryos for generation of stem cells. iPS cells can be potentially useful in pharmacologic-toxocologic in vitro studies to test drug sensitivity or to develop patient specific disease models. Furthermore, embryonic-like stem (i.e., iPS) cells open the possibility to use homologous recombination for gene therapy, thus avoiding insertional mutagenesis or long-term gene silencing. Already, one example of a successful gene repair strategy has been demonstrated by exchanging a defective hemoglobin gene in iPS to cure sickle-cell anemia in mice (24). Of course, the ultimate use of iPS may be in regenerative medicine (25, 26). Because iPS have infinite growth potential and allow vast in vitro expansion, the supply for tissue-specific cells should be unlimited. Replacement therapies with patient-derived tissues have the benefit of matching histocompatibility antigens and thus should avoid allogeneic T-cell responses and the need for immunosuppressive drugs (though natural killer cell responses and rejection owing to inflammation are other potential causes for transplant failures) (27). Numerous studies have already demonstrated that generation of beta-islet cells, myocardiocytes, dopamine-producing neurons or motor neurons may hold promise to help or to even one day cure such diseases as diabetes, myocardial infarction, neurodegenerative diseases or spinal cord injuries (28–33).
There are of course a number of hurdles to overcome before the use of iPS or ES cells for therapeutic purposes in patients comes to fruition, some of which can possibly be surmounted if we continue to study the molecular pathways of reprogramming and, in particular, chromatin patterns that are unique during differentiation. For example, the risk of tumorigenicity (by uncontrolled overexpression of transcription factors or by insertional mutagenesis) may be reduced if downstream events of de-differentiation are known and potential drugs could be developed that mimic the processes involved in reprogramming chromatin. Possibly, histone methyltransferases may promote the reprogramming event or circumvent the need for some transcription factors. ES cells pose the risk of teratoma formation; however, further detailed characterization of the pluripotent chromatin pattern should help to define reliable criteria for in vitro selection of lineage-specific cells, ensuring safe application in patients. Finally, the integration of tissue-specific cells in a functional organ context (for example, myocardiocytes form a syncytium with the myocardium to ensure synchronized contractions) will require further detailed knowledge of the epigenetic memory of lineage-specific cells. Thus, the bivalent chromatin pattern discovered by Bernstein and Lander leads to further studies––whether there are other unique chromatin patterns in pluripotent cells and whether distinctive enzymatic activities can be found acting exclusively in stem cells that could be manipulated––and raises the question of target specificity and how the bivalent marks are established at specific genomic locations.
Our view of differentiation as a one-way tour or down-hill slide has changed over time. We now know that differentiation is reversible and that it shows some characteristics of a see-saw: swinging easily back and forth between pluripotency and lineage specificity, conferring the benefit of using induced stem cells for therapeutic purposes but also the possible risk of tumorigenicity (Figure 1⇓). Recognizing key chromatin patterns for pluripotency is certainly one important first step in tipping the balance of differentiation in the future.
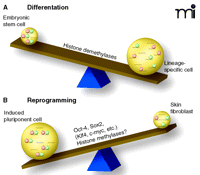
The see-saw of differentiation. Chromatin patterns of histone modification (i.e., acetylation, methylation, etc.) determine accessibility for transcription factors and, thus, direct tissue-specific transcription. A recent study identified a novel pattern of histone 3 methylation that is unique for embryonic stem cells (A). The epigenetic mark, termed bivalent [because it includes H3K4 methylation (green circles) as well as H3K27 methylation (pink circles)] can be found at the transcriptional start sites of many developmental genes (1, 2). These sites are poised for transcription in stem cells and resolve upon differentiation in either H3K4 or H3K27 methylation, associated with transcriptional activation or repression, respectively. Skin fibroblast cultures (upon introduction of four transcription factors) can revert into stem cells (iPS, induced pluripotent cells) (17). (B) Upon de-differentiation, the bivalent chromatin pattern is re-established. Identifying the key molecular mechanisms that are responsible for making the switch between pluripotency and lineage specificity opens the possibility of manipulating the see-saw of differentiation for regenerative medicine.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
-
↵1 Historically, SNF refers to the sucrose nonfermentable nucleosome remodeling complex identified in yeast.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Theresa Geiman, PhD, obtained her degree in Molecular and Cellular Oncology from The George Washington University. Her doctoral work focused on chromatin remodeling in development, using a mouse knockout model. Subsequent postdoctoral studies in Keith Robertson’s group at the NIH in Bethesda focused on DNA methyltransferase–protein complexes. Current work with Dr. Muegge at the NCI in Frederick involves epigenetic studies, emphasizing their effects in mammalian cellular differentiation, development, and cancer.

Sichuan Xi, MD, PhD, obtained his medical degree from the Binzhou Medical School, China, and his doctoral degree at The University of Hong Kong. As a research associate in University of Pittsburgh, he worked on the effects of epigenetic modification of TGFβ-EGFR-STAT signaling pathways in squamous cell carcinoma. Since joining Dr. Muegge’s lab, his research interests are to understand histone modification and regulation of chromatin structure in carcinogenesis and embryonic development.
Kathrin Muegge, MD, obtained her degree at the Medical School Hannover, Germany. During her postdoctoral work she studied cytokines and T-cell development, in the Laboratory of Molecular Immunoregulation at the National Cancer Institute. As a Principal Investigator in the Laboratory of Cancer Prevention at the National Cancer Institute in Frederick, she now investigates chromatin organization during embryonic development and in tumor progression. Her group has identified and cloned Lsh (Lymphoid specific helicase), a chromatin-remodeling protein that regulates DNA methylation patterns in mice. Using transgenic approaches, she and her colleagues are currently evaluating the molecular mechanism of cytosine methylation (and its interaction with histone modification pathways) and its role in gene expression during cellular differentiation and tumorigenesis. E-mail muegge{at}ncifcrf.gov; fax 301-846-7077.

