Cardiac Glycosides as Novel Cancer Therapeutic Agents
Abstract
The class of steroid-like compounds designated cardiac glycosides includes well-known drugs such as digoxin, digitoxin, and ouabain. Their continued efficacy in treatment of congestive heart failure and as anti-arrhythmic agents is well appreciated. Less well known, however, is the emerging role of this category of compounds in the prevention and/or treatment of proliferative diseases such as cancer. New findings within the past five years have revealed these compounds to be involved in complex cell-signal transduction mechanisms, resulting in selective control of human tumor but not normal cellular proliferation. As such, they represent a promising form of targeted cancer chemotherapy. New clinical studies of their anticancer potential as single or adjuvant treatments may provide insight into these potentially valuable therapeutic options. This review focuses on recent findings on cellular pharmacology of cardiac glycosides as they relate to treatment of human cancer and attempts to explain why these agents have been overlooked in the past.

History of the Use of Cardiac Glycosides for Cancer
The use of cardiac glycoside containing plants for medicinal purposes was first reported in ancient texts more than 1500 years ago. They have been used traditionally as arrow poisons, abortifacients, emetics, diuretics, and heart tonics. It is the latter pharmacologic activity that cardiac glycosides are most commonly associated with, and after 200 years, compounds such as digitalis and digoxin are still prescribed by Western doctors for control of congestive heart failure. Their use began after a meticulous analysis of a local herbalist’s formula in 1775 by the English physician and scientist William Withering. He found that a patient with “dropsy” (congestive heart failure) improved after administration of an extract containing foxglove (Digitalis purpurea L.) (1). Compounds extracted from foxglove and oleander include cardenolides (Figure 1⇓), such as digitalis, digoxin, and oleandrin, which increase cardiac contractility and act as antiarrythmic agents to control atrial fibrillation (2, 3). The mechanism of their action for the treatment of congestive heart failure arises from the inhibition of Na+,K+-ATPase, with a resulting increase in intracellular calcium concentrations. Cardiac glycosides, however, have a narrow therapeutic index, limiting their wider application to the treatment of other diseases, such as cancer.
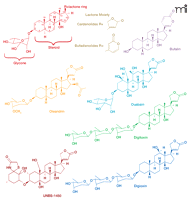
Structures of cardiac glycosides with antiproliferative activity. Representative cardenolide and bufadienolide cardiac glycoside compounds are presented.
Despite their potential to cause serious side effects, application of plant extracts containing cardiac glycosides for the treatment of malignant disease may extend back to Arab physicians in the eighth century (4). It is not just plants, however, that contribute to our appreciation of cardiac glycosides possibly having a role in cancer management. An ancient Chinese medicinal treatment of cancer still in use today involves application of an extract of secretions of the Bufo bufo toad, known to contain multiple bufodeninolides (another type of cardiac glycoside), including bufalin (Figure 1⇑) (5–7). The potential use of cardenolide-like compounds for the treatment of cancer, initially investigated forty years ago, was abandoned because of the toxicity of these compounds (8, 9). It was only recently, however, that Scandinavian oncologists suggested that the apoptosis produced by digitalis in human tumor cells occurred at concentrations that could be achieved without toxicity in humans and, therefore, this agent might be useful for treatment of cancer (10–12). In 1979, Stenkvist et al. (13) noted that the altered morphology of breast cancer cells from women treated with digitalis (who had undergone mastectomy). Women receiving digitalis had tumor cells with more benign characteristics than those tumor cells in patients not receiving this cardiac glycoside. Moreover, the cancer recurrence rate of women taking digitalis was lower, suggesting an important beneficial anticancer effect of this cardiac glycoside (14).
Within the past ten years, there has been a substantial increase in the number of studies observing the effects of cardiac glycosides on the growth of human malignant tumor cells. A review of the literature indicates a surprising variety of plants and even animals whose extracts and isolated cardiac glycoside compounds have been cited for their antiproliferative effects (Table 1⇓). The purpose of the present review, therefore, is to examine the hypothesis already expressed by some (10, 14–20), that use of selected cardiac glycosides may represent a worthwhile approach toward control of malignant cell proliferation even despite their narrow therapeutic index. This is all the more timely because promising clinical trials of cardiac glycosides and extracts containing them have recently been initiated.
List of Plants and Animals with Cardiac Glycosides Having Antiproliferative Activities
Na+,K+-ATPase: Beyond Cell Membrane Exchange OF Na+ AND K+
Na+,K+-ATPase, as an energy-transducing ion pump, has been studied extensively since its discovery in 1957 (21). This enzyme consists of two types of subunits, designated α and β , in addition to a single-transmembrane-spanning protein, FXYD––named for the conserved amino acids in its signature motif: (Phe-Xxx-Tyr-Asp). The α subunit, responsible for binding of Mg2+, ATP, Na+, K+, and cardiac glycosides, is considered the catalytic subunit of the enzyme. The β subunit is a glycoprotein that seems to act as an adhesion molecule that regulates gap junction proteins; is involved in structural and functional maturation of the holoenzyme; facilitates transport of the α subunit to the plasma membrane and maintenance of the enzyme in the lateral membrane of epithelial cells (15–17). The function of the FXYD protein involves regulation of the enzyme function, thus adapting the kinetic properties of active Na+ and K+ transport to the specific needs of different cells (22, 23). Four α subunit variants, as well as three β , and seven FXYD subunit variants have been identified (17). The well-established function of Na+,K+-ATPase is to use ATP as an energy source to drive excess Na+ out of cells in exchange for K+, thereby maintaining an essential ionic and osmotic balance. Binding of certain α subunits by cardiac glycosides inhibits ATP binding and disrupts the ability of the enzyme to perform this exchange in an efficient manner. This, in turn, results in an enhanced entry of calcium into cells, which, in the event of failing cardiac myofibrils, helps produce a more efficient myocardial contraction and improves cardiac pump activity.
What, then, is the evidence supporting the hypothesis that Na+,K+-ATPase may be an important target for cancer therapy? For the past ten years, published studies have suggested a role for Na+,K+-ATPase in regulation of cell growth and expression of various genes beyond that of ion transport. Davies et al., for example, observed altered Na+,K+-ATPase activity in premalignant mucosa months before tumor development induced by the carcinogen 1,2-dimethyldrazine (24). There have also been reports of increased expression of particular subunits of Na+,K+-ATPase in gastric (25) and bladder cancers (26). In addition, alterations in overall Na+,K+-ATPase activity and relative subunit abundance were observed in a highly invasive form of human renal carcinoma cells (27), non-small cell lung cancer (28), and carcinoma cell lines obtained from a number of other tissues (29). It would appear, however, that simply looking at enzyme subunit content or relative activity in malignant and non-malignant tissue may not provide adequate insight into the role of this enzyme in cancer. This can now be interpreted in the light of newly proposed consequences of cardiac glycoside binding to Na+,K+-ATPase.
Proposed Mechanism(s) OF Cardiac Glycoside–Mediated Antiproliferative Effects
An explanation of the role of Na+,K+-ATPase in complex cell signaling pathways, many of which are of critical importance to malignant cell proliferation, has been put forth by Xie and colleagues (30–32). They have shown that binding of cardiac glycosides (e.g., ouabain) to Na+,K+-ATPase triggers a complex signaling cascade that is initiated by interacting with neighboring membrane proteins and organized cytosolic cascades of signaling molecules. These signaling complexes send messages to intracellular organelles via the activation of the protein tyrosine kinase Src, transactivation of epidermal growth factor receptor (EGFR) by Src, activation of Ras and the p42/44 mitogen-activated protein kinases [MAPKs, also termed extracellular-regulated protein kinases 1 and 2 (ERK1/2)], and increased generation of reactive oxygen species (ROS) by mitochondria. Activation of these cellular pathways is also linked with translocation of Na+,K+-ATPase, through endocytosis, to the nucleus (17). In fact, Xie and colleagues (17, 30) have referred to this as the Na+,K+-ATPase-Src-caveolin “signalosome” complex (Figure 2⇓). The view of Na+,K+-ATPase as a simple ion-exchange pump situated solely at the cell membrane is thus outmoded. Further research on this enzyme, including its role in regulation of cell proliferation and its inhibition through cardiac glycosides, is clearly warranted.
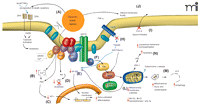
The Na+,K+-ATPase signalosome complex. The binding of selected cardiac glycosides (CGs)––such as oleandrin, bufalin, and digitoxin––to Na+,K+-ATPase results in complex but well-documented changes in cell signaling events. The “signalosome” complex includes the enzyme, Na+,K+-ATPase as well as Src, phosphoinositide-3 kinase (PI3K), and phospholipase C each of which, in turn, sets into action complex signaling events that can result in tumor cell death through either apoptosis or autophagy-related mechanisms. Administration of CGs can (A) increase the (cell surface) expression of death receptors (DR4, DR5) and activate caspase activity; (B) result in increased intracellular calcium concentrations, which, in turn, (C) decreases the expression of transcription factors such as Activator Protein-1 (AP-1). CG treatment can also (D) inhibit activation (i.e., block the phosphorylation) of Akt, which normally blocks apoptosis; (E) inhibit activation of the transcription factor Nuclear Factor-kappaB (NF-κ B); (F) activate the Ras pathway, leading to increase activity of Raf–MAPK pathway; (G) activate Src; (H) inhibit tumor necrosis factor (TNF)-mediated activation of NF-κ B by inhibiting the binding of tumor necrosis factor receptor 1–associated death domain protein (TRADD) to the cellular membrane; (I) inhibit extracellular transport of tumor growth factors, such as fibroblast growth factor-2 (FGF-2); (J) alter membrane fluidity which, in turn, may inhibit Fas-related signaling; (K) lead to the production of reactive oxygen species (ROS) with subsequent injury to mitochondria; (L) produce a decrease in mitochondria membrane potential and a decrease in quantity of anti-apoptotic proteins Bcl-XL and Bcl-2 and topoisomerases I and II; and (M) cause mitochondrial condensation and loss of function, that, in turn, can lead to autophagic processes and cell death (N). Adapted from (30).
The diverse mechanisms reported to specifically be involved in cardiac glycoside-mediated control of malignant cell proliferation has been compiled (Table 2⇓ and Figure 2⇑), and there are several excellent reviews on this subject (15–17). There are unifying themes that link mechanisms involving the water-soluble (ouabain) and relatively lipid-soluble (oleandrin, bufalin, and digitoxin) cardiac glycosides, including activation of ERK1/2; increased expression of the cell cycle inhibitor p21Cip1 and consequent inhibition of cell cycle progression (through decreased expression of cyclin proteins); inhibition of transcription factors, such as Nuclear Factor-kappaB (NF-κ B) and Activator Protein-1 (AP-1); inhibition of Akt (a protein serine–threonine kinase) and related critical components of the phosphoinositide-3 kinase (PI3K) pathway; initiation of death receptor–mediated apoptosis; sustained ROS production with consequent mitochondrial injury; and inhibition of topoisomerases and reduction in expression of anti-apoptotic proteins, such as Bcl-xL and Bcl-2. Although the known primary target of cardiac glycosides is Na+,K+-ATPase, not all of the reported mechanisms of antiproliferative action (Table 2⇓) are related to inhibition of this important enzyme. That is, cardiac glycosides such as bufalin and oleandrin should be thought of as pleiotropic or multi-mechanistic anticancer agents. Few, if any, of these published reports on mechanism of action of cardiac glycosides, however, address reasons for the well-established differential in cardiac glycoside-mediated effects on human- vs rodent-derived malignant cell lines.
Reported Mechanisms of Cardiac Glycoside–Mediated Inhibition of Tumor Cell Proliferation
Species-Dependent Sensitivity AND Selective Human Tumor Cell Response TO Cardiac Glycosides: Important Mechanistic Clues
An unusual attribute of compounds such as oleandrin, bufalin, and digitoxin is that they are, on the one hand, almost completely nontoxic to rodent (mouse, rat, and hamster)-derived tumor cell lines but potently inhibit proliferation of monkey and human tumor cell lines at nanomolar concentrations (33–36). These results have been confirmed across a wide spectrum of human and rodent tumor cell lines, including those of hematologic and solid tumor derivation. This species-dependent disparity in tumor cell sensitivity to a proposed antitumor agent is unusual. It may, in fact, have contributed to the conclusion in the 1970s that cardiac glycosides were without efficacy because murine P388 and L1210 lymphocytic leukemia cell lines were, at the time, the only cell lines available for anticancer drug development. The magnitude of difference in response of murine as compared to human tumor cell lines suggests that a fundamental difference in drug targeting exists and, thus, serves as a probe or model for re-examination of the pharmacologic role of this class of compounds in treatment of human cancers.
Whereas the differences between human and murine responses to cardiac glycosides are interesting and should be studied further, the differential effects of cardiac glycosieds on human tumor vs normal cells are essential to their usefulness as a therapy. Several published studies have confirmed the observation that cardiac glycosides have a selective effect on malignant but not normal cell proliferation. For example, oleandrin suppresses the activation of certain transcription factors and potentiates ceramide-induced apoptosis in human tumor cells but not in normal, primary human cells (19). In vitro observations that leukemia cells undergo apoptosis in the presence of oleandrin and bufalin, but that normal leukocytes do not, are also consistent with the hypothesis of a potentially therapeutic, selective therapeutic effect of cardiac glycosides on tumor growth (37–39). Not only do cardiac glycosides appear to be more effective at inhibiting proliferation of malignant cells than normal cells, but they also are more effective at sensitizing tumor cells to irradiation, which would appear to increase their potential utility in the clinic. Research reported by several investigators (40–42) indicates that cardiac glycosides sensitize human tumor but not normal cells to subsequent radiation treatment. These data suggest that it may be possible to exploit differences in the Na+,K+-ATPase pumps of normal as opposed to tumor cells to improve the therapeutic index of radiation therapy.
Modern drug development seeks specific biochemical differences between malignant and normal cells that may be critical to survival of cancer cells. One then attempts to develop selective inhibitors to disrupt these pathways. Na+,K+-ATPase, however, is a ubiquitous enzyme present in every mammalian cell. At first appearance, therefore, it would appear to make Na+,K+-ATPase an anticancer target of dubious value unless, of course, the target were found to be fundamentally different in normal versus malignant human cells or between rodent and mammalian cancer cells. Our recent data suggest that, in fact, there is a difference in the basic subunit composition of Na+,K+-ATPase that might explain the differential species-dependent sensitivity to cardiac glycosides. Although human tumor cells and tissues commonly express both α 1 and α 3 subunits, all rodent tumor cell lines we have examined to date only express the α 1 subunit (Figure 3⇓). Early reviews of the biochemical properties of Na+,K+-ATPase suggested that cardiac glycoside binding may be equal to all four α subunit isoforms; however, more recent studies have shown a clear preferential binding of cardenolides to the α 3 form over that of the α 1 or α 2 isoforms (43–45). For example, O’Brien et al. (45) cite a 1000-fold difference in binding of ouabain to the α 3 isoform over that of α 1. Given the fact that rodent tumor cells possess the α 1 subunit, lack expression of α 3, and are unresponsive to inhibition of proliferation with cardiac glycosides, we suggest that the α 3 subunit is critical. The increased expression of α 3 over α 1 subunits has also been noted in human colon colorectal cancer and colon adenocarcinoma cell lines (e.g., KM12-L4, T-84, HT-29, and WiDr), whereas no significant expression of the α 3 isoform protein was noted in the normal kidney and renal tissues (46). Moreover, human tumor cell lines with a low ratio of α 3:α 1 are relatively resistant to growth inhibition with cardiac glycosides but those tumor cell lines with high α 3:α 1 ratios are very sensitive (Figures 3⇓ and 4⇓). This finding, of course, also suggests that determination of the relative α 3:α 1 ratio in tumor biopsy specimens may have some prognostic value if that patient is to be subsequently treated with a cardiac glycoside.

Relative human and rodent tumor cell sensitivity to oleandrin correlates with Na+,K+-ATPase subunit composition. A. Mouse pancreatic cancer cells that lack expression of the α-3 subunit (Panc-02) are non-responsive to oleandrin whereas human tumor cell lines MiaPaca and Panc-1 (that contain high expression of α-3 relative to α-1) are extremely responsive. The human pancreatic tumor cell line BXPC3 that contains a very low level of the α-3 subunit, as per immunoblot analysis (B), is somewhat resistant to cytotoxic effects of oleandrin. These data suggest that it is the lack of α 3 in rodent tumor cell lines that explains their resistance to cardiac glycosides. In addition, the data suggest that it is the relative α 3:α 1 ratio correlates with human tumor cell sensitivity to lipid soluble cardiac glycosides such as oleandrin.
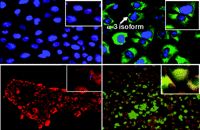
Relationship of expression of Na+,K+-ATPase α 3 subunit and binding of oleandrin to human pancreatic cancer cell membranes. Cells were stained using a mitochondrial dye, Mitotracker, (red color) and a nuclear Herscht dye (blue). Top row: Cells were also exposed to an antibody to the α 3 subunit of Na+,K+-ATPase (green color). Bottom row: Cells were incubated with a fluorescent analog of oleandrin (green color). The presence of green staining (top row) indicates the relative presence of the α 3 subunit which is more prevalent in the PANC-1 than the BXPC3 cells. There is a corresponding uptake of oleandrin (bottom row) only in those cells (PANC-1) that express the α 3 subunit.
Mijatovic et al., on the other hand, suggest that, rather than the α 3 subunit, it is the α 1 subunit of Na+,K+-ATPase that could represent a novel anticancer target (47). They have shown that human lung cancer cell lines overexpressing the α 1 subunit were sensitive to a few select cardenolides. They noted that the cardiac glycosides produced a marked change in the actin cytoskeleton, suggesting this abets tumor cell death. Whether it is altered expression of α 1, as suggested by Mijatovic et al., or an elevation of α 3, as indicated by our own work, or perhaps even a specific ratio of α 3:α 1 that is most important as a predictor of cell sensitivity, remains to be determined. More research, using human tissues and not just cell lines, will no doubt shed light on the potential importance and perhaps even prognostic value of the enzyme subunit composition within individual types of tumors.
It is significant that the relative composition of Na+,K+-ATPase subunits may not be static within human tissues. The relative ratio of α subunits within the enzyme may shift when tissues are transformed from a benign to a malignant state. Sakai et al. (46) recently showed, for example, that a decrease in the α 1 isoform and an increase in the α 3 subunit occurs in colon tissue when a normal phenotype changes to a malignant one. If, as our data suggest, it is the relative expression of α 3 that is important for determining sensitivity of a tissue to inhibition by cardiac glycosides then, in essence, the report by Sakai et al. suggests that the tumor becomes a more sensitive target than normal tissue to cardiac glycoside therapy (46). Given the current as well as proposed clinical trials of cardiac glycosides for treatment of cancer, specific determination of enzyme subunit composition in specific tissue types as well as pathologic characterization may prove to be a timely tool to help optimize the effectiveness of this class of potential cancer therapeutic agents.
Cardiac Glycoside-Mediated Cancer Cell Death: Autophagy AND Apoptosis
Although it is clear that lipid-soluble cardiac glycosides (i.e., digitoxin, oleandrin, and bufalin) have a potent ability to produce human tumor cell death, the mechanisms by which this is accomplished are still being defined. Apoptotic cell death mediated by cardenolides has been demonstrated in a number of cell lines. Sreenivasan et al., for example, have shown that oleandrin produced an increase in expression of Fas and Tumor Necrosis Factor Receptor 1 (TNFR1), resulting in potentiation of apoptosis in tumor cells but not in normal primary cells, such as peripheral blood mononuclear cells or neutrophils (48). Fas–Fas ligand and TNF–TNFR1 death pathways are important mediators of apoptosis (49). Another recent report has shown that oleandrin, bufalin, digoxin, and digitoxin initiate apoptosis induced by Apo2L/TNF-related apoptosis-inducing ligand (TRAIL) in non-small-cell lung cancer cells by increasing the expression of death receptors 4 and 5 (18). Because Apo2L/TRAIL induces apoptosis in tumor cells with little if any toxicity to normal cells, this cytokine is of great interest to cancer researchers. The selective cardenolide activation of death receptors may very well contribute to the observation that compounds such as oleandrin are relatively selective in their cytotoxic activity.
Oleandrin elicits caspase-associated apoptosis in human prostate carcinoma cells (50). Interestingly, however, treatment of human PANC-1 pancreatic cancer cells produces clear hallmarks of autophagy, including formation of autophagosome bodies with damaged mitochondria and expression of light chain-1 protein, an early indicator of autophagosome formation (51). Frese et al. have also suggested that the apoptotic potential of cardiac glycosides depends on the cell type treated (18). Our data on the differential effects of oleandrin on tumor cells, such as pancreatic vs prostate tumor cells, as compared to oleandrin-treated normal human cells concurs with this.
Cardiac Glycosides and Estrogen Receptor Interaction
Selected cardiac glycosides may be of particular importance in the treatment of human breast cancer. Chen et al. (15) have recently suggested several reasons why cardenolides should be developed as anti-breast cancer drugs. These include the facts that: 1) Na+,K+ -ATPase is a key player of cell adhesion and is involved in cancer progression; 2) the enzyme serves as a versatile signal transducer involving a number of hormones, including estrogens; and 3) the aberrant expression and activity of this enzyme in breast cancer implicates an etiologic or at least contributing role of Na+,K+-ATPase in the development and progression of this malignant disease. For example, there is now strong evidence that Na+,K+-ATPase plays an important role in the assembly of tight junctions (TJs) and cell adhesion (52–55). Chen et al. convincingly argue that altered expression and malfunction of Na+,K+-ATPase may lead to abnormal TJ structure and, thus, to altered cell adhesion important in the progression of breast cancer. As mentioned previously, there are also strong data supporting the role of Na+,K+-ATPase in a complex signalosome involved in transmitting membrane signals to the nucleus.
A series of reports suggests that estrogen receptor (ER) ligands (e.g., 17β-estradiol and estrogen-like molecules) can also serve as ligands of Na+,K+-ATPase. Use of 17β-estradiol enhances Na+,K+-ATPase activity (56) possibly through improvement of the interaction of the enzyme with ATP as well as Na+ and K+ ions. Because the interaction of 17β-estradiol with ERs serves as an important determinant of breast cancer growth, and cardiac glycosides can block this interaction, cardenolides could be considered effective modulators of estradiol-dependent breast cancer proliferation.
Cardiac Glycosides and Cancer Prevention
Recent investigations of potent cardiac glycosides have focused on their potential application to the treatment of established cancers; however, at least one report has suggested that there may also be a chemopreventive role for this class of agents. That is, Afaq et al. have suggested that oleandrin might serve as an effective agent for the prevention or treatment of skin cancer (57). Their research investigated the topical application of oleandrin to CD-1 mice to counteract the effects of TPA (12-0-tetradecanoylphorbol-13-acetate), a widely used skin tumor promoter. The topical application of TPA to mouse skin or its treatment in certain epidermal cells is known to result in several biochemical alterations, changes in cellular functions, and histological changes leading to dermal tumor promotion. The data of Afaq et al. clearly show that application of oleandrin to skin prior to TPA administration affords significant inhibition of TPA-induced skin edema, hyperplasia, epidermal ornithine decarboxylase (ODC) activity, and protein expression of ODC and cyclooxygenase-2 (COX-2), classical markers of inflammation and tumor promotion. Their data also show that topical application of oleandrin prior to TPA inhibits activation of PI3K and phosphorylation of Akt, activation of NF-κ B, and degradation and phosphorylation of the inhibitor of NF-κ B α protein (Iκ Bα ). These authors, therefore, recommend the use of chemopreventive agents (i.e., oleandrin) in formulations such as emollients or patches for the prevention or treatment of skin cancer (57). This suggestion is all the more relevant when considered in light of our own work which shows a potent ability of oleandrin to inhibit human melanoma proliferation (58).
In Vivo Efficacy AND Development OF Cardiac Glycosides for Clinical Cancer Therapy
Rodent tumor cells fail to respond to cardiac glycosides in vitro. Similarly, it has been very difficult to demonstrate an in vivo response of syngeneic rodent tumors to administration of this class of compounds. However, as shown in Figure 4⇑, there is no question that human tumor cell lines are extremely sensitive to treatment with cardiac glycosides such as oleandrin and bufalin. Thus, it is possible that human tumor xenografts would respond. Indeed, this is exactly the case, as shown by several investigators. Han et al. (7), for example, explored the response of a human hepatocellular carcinoma cell line (BEL-7402) implanted orthotopically (i.e., transplantation of cells or tissue into its normal anatomical site) in liver tissue to intraperitoneal treatment with bufalin. They found that this toad-derived cardiac glycoside produced significant reductions in tumor volumes and a prolongation in life-span of the animals. Importantly, no adverse morphological changes were noted in myocardial, hepatic, or renal tissues. Another interesting report involved use of the semi-synthetic cardenolide UNBS-1450 against orthotopically implanted human non-small-cell lung cancer (59). Chronic oral administration of this compound produced a beneficial therapeutic effect against these orthotopically implanted A549 tumor cells. Finally, digoxin inhibits human neuroblastoma growth in vitro and significantly reduced the growth of human neuroblastoma tumor in vivo in mice (60), whereby the antitumor effect arose, at least in part, from an antiangiogenic effect because digoxin was also found to be effective in the chicken chorioallantoic membrane (CAM) assay. Thus, although limited, evidence exists for antitumor activity against human tumor xenografts.
In this age of targeted therapeutics, however, where one strives for selective effects within tumor cells so as to increase effectiveness and also minimize toxicity to normal tissues, one might reasonably question why cardiac glycosides with a known narrow therapeutic index would ever be considered for clinical development as an anticancer therapy. There is little doubt that the potential for serious cardiovascular toxicity exists with many, if not all cardiac glycosides, but the risks appear manageable because the effective concentrations (i.e., in the nanomolar range) of these agents to control cancer-cell proliferation are well below concentrations that produce cardiac toxicity (61). Because the cardiovascular toxicities associated with this class of agents are well documented, careful monitoring of plasma concentrations may allow for the continued safe use of digitalis and related compounds. Moreover, specific antibody-based treatments, such as digoxin-specific Fab antibody fragments (Digibind; DigiTAb), as well as an older treatment, activated charcoal, are available to rescue patients receiving accidental over-medication (62, 63).
The desirable goal of producing a synthetically derived cardiac glycoside with potent ability to inhibit proliferation of human tumors but without the potential to cause cardiac related toxicity, unfortunately, has not yet been fully achieved. Investigators have shown, however, that single cardenolides, or those derived from various extracts of plants and animals (Table 1⇑), represent potent compounds with selective effects against human tumor cell lines and xenografts. For example, UNBS-1450, a semisynthetic cardenolide derivative of a cardiac glycoside originally isolated from an African plant (Calotropis procera), has shown promising activity against human non-small cell lung carcinomas growing as xenografts in nude mice (59). UNBS-1450 can be safely administered to mice in doses that are twenty-four times greater than that of ouabain and twelves times greater than that of the parent compound oxovoruscharin (64). With good activity against non-small cell lung cancers in mice with metastases in the brain and liver, UNBS-1450 is entering phase I clinical trials in Belgium (17).
To date, there is only a single plant extract, Anvirzel™, derived from Nerium oleander, that has progressed through a Phase I trial in the United States. Anvirzel is a hot-water extract of oleander whose encouraging preclinical activity (65) led to a successful investigational new drug application (IND) from the FDA and to a Phase I clinical trial performed at the Cleveland Clinic between 2000 and 2001. No objective responses were noted which might be because the limited time of exposure to the product and intramuscular route of administration limited the total volume of extract that could be administered on a daily basis. A longer time of exposure and a different route of administration may impact response. Also, no dose-limiting toxicities were found. The product known as PBI-05204 was produced in response to the need for a formulation and route of administration suitable for adequately exploring the anticancer potential of an oleander extract. PBI-05204 is a modified supercritical CO2 extract of organically grown Nerium oleander that has been especially formulated for oral administration to humans. An IND for evaluation of PBI-05204 as a “botanical drug” was obtained from the FDA in September, 2007, and a Phase I clinical trial in patients with solid tumors has now been initiated at the University of Texas M.D. Anderson Cancer Center. Given the expanding knowledge of the multiple anticancer mechanisms for cardenolides, the outcome of these early stage clinical trials of potent plant extracts, such as the proposed botanical drug PBI-05204 and simpler single chemical entities such as UNBS-1450, will be of great interest. The results may hold the key as to whether this class of potent natural products is worthy of further development as primary and/or adjuvant therapy for malignant diseases.
Conclusions
Our understanding of the spectrum of the pharmacologic activities of cardiac glycosides has increased significantly since the discovery of their effectiveness for treatment of congestive heart failure. It is now recognized that certain cardiac glycosides are involved in complex cell signal transduction mechanisms that may have important consequences in their application to the prevention and/or treatment of malignant diseases. Development of clinically targeted, antiproliferative cardiac glycosides could be helped by systematic evaluations of several formulations and chemical variants. Furthermore, assays of the relative presence of α 1 and α 3 subunits in clinical samples, for example, may give some direction for the assessment of which cancers might be most susceptible to cardiac glycoside therapy. Further development of synthetic, semi-synthetic, or naturally occurring cardiac glycosides, with assessment of their toxicity and structure-activity relationships, might expand the possibilities of finding a cardiac glycoside with a wider therapeutic index. Because of concerns with toxicity of internally used cardiac glycosides, topical formulations should also be considered for skin cancer prevention and/or treatment. Additionally, because chemotherapy has had limited benefits in most advanced malignancies, cardiac glycosides could also be investigated for possible adjuvant therapy. They may, for example, be of benefit in early-stage disease as well as stand-alone therapy for patients where conventional interventions are either not applicable, because of patient tolerance, tumor cell sensitivity, or where a patient has run out of conventional therapeutic options altogether. Finally, consideration should be given to the use of selected cardiac glycosides or natural extracts containing them as adjuvant therapy for concomitant use with currently existing therapies. Given that certain Na+,K+-ATPase subunits appear to make unique targets that are selectively expressed in tumor as opposed to normal tissue and that the potential for cardiac toxicity is not shared by many other currently available cancer therapies, cardiac glycosides should be considered as an therapeutic option for novel combination therapy.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Alison Pawlus, RPh, PhD, completed her graduate training, in 2007, in Pharmacognosy at the University of Illinois at Chicago in the laboratory of Dr. A. Douglas Kinghorn. She is a Postdoctoral Fellow in the Department of Experimental Therapeutics at the University of Texas, M.D. Anderson Cancer Center where she studies the use of natural products for cancer treatment and prevention.

Peiying Yang, PhD, (Collaborator) is an Assistant professor in the Department of Experimental Therapeutics. Dr. Yang’s research has focused on bioactive lipids and natural products in cancer development and prevention. She developed a rapid, specific, and sensitive method for simultaneously determination of arachidonate metabolites in various biological matrices. Additionally, Dr. Yang is interested in the effects of nutritional supplements, such as fish oil, and Chinese herbal medicine.

Keith I. Block, MD, co-founded and is Medical/Scientific Director of the Block Center for Integrative Cancer Treatment. He is a member of the National Cancer Institute’s Physician Data Query (PDQ) Cancer CAM Editorial Board, the editor-in-chief of the peer-reviewed journal Integrative Cancer Therapies, and a Clinical Assistant Professor at the College of Medicine at the University of Illinois, Chicago. In collaboration with the University of Illinois and other university facilities in the US and Israel, Dr. Block conducts research in nutrition and in the use of natural medicines in cancer treatment.

Robert A. Newman, PhD, received his undergraduate training at the University of Rhode Island and graduate training at the University of Connecticut, where he obtained MS and PhD degrees in pharmacology and toxicology. His postgraduate work was performed at the Medical School of Georgia and at the University of Vermont Medical School. After a sabbatical at Stanford University in 1983, Dr. Newman joined the faculty of the University of Texas M.D. Anderson Cancer Center. He is a Professor of Experimental Therapeutics and holds the D.B. Land Professorship. Dr. Newman also jointly runs the Pharmacology and Analytical Core lab for the institution as well as co-directs the Pharmaceutical Development Center that has introduced more than six compounds into the clinic over the past seven years. He is the author of over 250 peer-reviewed publications and several books. His current research deals with the science of nutraceuticals and understanding how these can be specifically applied for prevention and treatment of inflammation and malignant disease. E-mail rnewman{at}mdanderson.org; fax (713) 563-9093.

