Rear-view Mirrors and Crystal Balls: A Brief Reflection on Drug Discovery
- John S. Lazo, PhD
Now in its centennial year, ASPET arguably faces the most interesting period that has ever existed for pharmacology and drug discovery. The genomic sequences of hundreds of species, including our own, offer a profound view of who we are, how we evolved, and the nature of our microscopic enemies. Moreover, genomic advances afford us with a heretofore unimaginable number of new molecular therapeutic targets. Novel methods in synthetic bioorganic and medicinal chemistry, and innovative approaches in computational chemistry, likewise promise profound changes in drug discovery paradigms. All of these advances are a product of the last half-century of public and private investment in biomedical research. ASPET’s Centennial celebration this year inspires us to reflect on the history of the drug discovery process, and also motivates us to consider the anticipated seismic changes in drug discovery tools. What are the lessons learned, and how might they affect the future? Undertaking the task of outlining even a limited history of drug discovery would obviously be daunting. But as we enter into ASPET’s second century, examination of broad issues of drug discovery, however subjective, will continue to shape our discipline and the Society itself.
The Foundation of Drug Discovery
Although it is common to portray drug birthing as a linear process, starting with a discovery phase that burgeons into a development phase, it should be obvious that in reality the process is rarely linear. Indeed, there are many roads to a successful new therapy, which usually entail frequent intellectual, scientific, practical, and commercial twists and turns. In the ancient times, human remedies were largely derived from extracts of local plants, animals, or minerals. Even today in some cultures traditional medicines remain a central component of the health care system. In developed nations, herbal remedies are becoming increasingly popular.
In the sixteenth century, Paracelsus urged alchemists to extract the pharmacological essence of mixtures, but it was the innovative chemists of the nineteenth century that finally began to systematically isolate and define pharmacologically active substances. Perhaps the most profound modifier of therapeutics in the western world was the establishment of an institute of pharmacology, at the University of Strasbourg, under the leadership of Oswald Schmiedeberg (1). But beyond the institutionalization of the new discipline, substantial developments in new synthetic and analytical chemical methodologies, especially the emerging dye industry, were widely influential. The new dyes invigorated chemistry and provided the fundamental reagents that inspired Paul Ehrlich to posit the existence of “chemoreceptors,” a critical concept in the evolution of molecular targets for drugs.
Progress in chemistry has continually fueled the creation of new drugs (Figure 1⇓). Admittedly, serendipity has always played a role in science, and the invention of aspirin, still the most widely used drug in the world, was certainly an example of this fact in drug discovery. Nonetheless, the popular adoption of this seminal nonsteroidal anti-inflammatory agent encouraged others to discover and develop the wonder drugs of the early twentieth century, including the sulfa drugs. And fortune indeed favors the prepared mind; the maturation of other scientific disciplines reflects the intellectual power that has fueled progress in rational pharmacology. For example, penicillin would not have been discovered without the microbiological advances in the laboratories of Alexander Fleming and others. Other disciplines during the last century, including physiology, biochemistry, microbiology, and molecular biology, are similar bricks in the foundation of drug discovery [for example, see (2–4)].
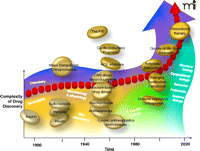
Timeline of Drug Discovery. Some of the major classes of drugs discovered and developed in different periods are noted. The dominant influential disciplines affecting the drug discovery and development process are also shown.
Communication and collaboration across scientific disciplines has enhanced the complexity of the drug discovery and development process. Moreover, many diseases that are now recognized to be therapeutically attractive are polygenic or affected by redundant pathways. Thus, identifying a compound that acts on a single molecular target is often inadequate. Patients frequently require several drugs concurrently, and one must be concerned about overlapping toxicities, common metabolic pathways, and other drug-drug interactions. These concerns are confounded by pressures to circumvent antibiotic, antiparasitic, or anticancer drug resistance mechanisms. The causes of drug resistance are complex, but the consequences are clear. The life cycle of effective drugs is reduced, and newer drugs must be used all the more sparingly, which constrains commercial interests and incentives (5). The chemical nature of drugs themselves also grows more complex for example, the macromolecular dimensions of biologics posing new challenges in terms of production, drug delivery, and administration. The success rate from first-in-human to registration remains remarkably low (approximately ten percent), illustrating how difficult it is to predict which candidate compound will be clinically successful (6, 7). The number and types of compounds to be investigated continues to expand because of new combinatorial and diversity-oriented synthesis methodologies, offering both challenges and opportunities. There are now millions of compounds that are commercially available and can be interrogated.

Strategies for Identifying Drugs— and Drug Targets
The search for new pharmacological agents historically has followed two general pathways: forward pharmacology and reverse pharmacology (Figure 2⇓). As illustrated in Figure 1⇑, the early days of discovery and development of new pharmaceutical agents was dominated by empirical approaches that capitalized on enlightened serendipity. Potential drugs were identified using physiological methods with animals, isolated organs or tissue and bacterial preparations. This forward pharmacology approach had the distinct advantage of involving compounds that acted in vivo or in situ, enhancing the likelihood of achieving the desired physiological effect in humans, and required little or no understanding of the mechanisms of action of the compound. If an animal model was used, there was the added benefit that the investigator might be able to deduce some information about the duration of action, safety, and possibly metabolism of the drug. The approach was unbiased in that the investigator had no preconceived molecular target. On the other hand, in those instances where good animal models did not exist, forward pharmacology was not possible.
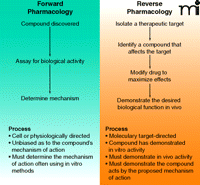
The Two Fundamental Approaches to Drug Discovery.
The alternative approach, which has become increasingly popular with the advent and growth of biochemical and molecular biological tools, is reverse pharmacology. This highly reductionist approach relies on the isolation of an identified molecular target, often an enzyme, and pursuit of a small molecule to modulate the target’s known or predicted function. Small-molecule inhibitors of enzyme function are generally more attractive than biological activators because of the belief that gain-of-function is a much more energetically challenging task than is loss-of-function. Nonetheless, we have recognized for some time that the highly complex signaling pathways permit indirect attacks on even gain-of-function defects (8), including examples of small molecules or stapled peptides that can activate defective proteins [e.g., p53 (9, 10)]. Highly simplified test systems have been designed to probe the nature and kinetics of small-molecule inhibition. Computational methods and the in silico investigation of pharmacological agents at target sites are now complemented by high-resolution crystal and NMR structures. These and other avenues make reverse pharmacology intellectually compelling, although compounds identified with simplified in vitro assays may be unable to reach the target in vivo, or may otherwise reveal “off-target” effects. In contrast to forward pharmacology, where deconvolution of compound activity in the animal test system is the critical step, reverse pharmacology faces the hurdles of clear in vivo activity and appropriate target modification. The identification of the target in reverse pharmacology is itself multifaceted and must be considered in the context of signaling pathways (Figure 3⇓).
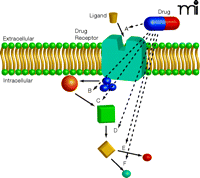
Potential Targeting of Receptors and Signaling Pathways. Although we often think of drugs in terms of pathway A, where the drug blocks the ligand interaction with the receptor, many different sites of action can be considered. Some targets will be more “druggable” than others, which may assist in refining the approach for any particular disease process. In many situations, however, the detailed kinetics of the pathway and the collateral signaling effectors are unknown, making a decision problematic. In some cases, it may be desirable to disrupt the pathway at several levels to ensure effective disruption and to reduce the likelihood of pathway disruption compensation or drug resistance. In other cases, disruption of one pathway may lead to unwanted toxicities.
Historically, since Erhlich’s chemoreceptor concept, drugs have been developed to block receptors and/or enzymes. Left untouched have been the thousands of transient, therapeutically interesting, protein protein interactions. Protein protein interfaces have been considered flat and relatively featureless, particularly when compared to the deep active sites of enzymes or receptor binding sites. The large surface areas of such interactions often 1,200–4,000 square angstroms have made it difficult to imagine how a relatively small molecule could ever effectively disrupt the interactions. Nevertheless, mutations of single amino acids reveal surface hotspots, amenable to intervention, at topologically attractive regions (11), and the discovery of several compounds that effectively disrupt protein protein interactions has further stimulated this approach. Fragment-based chemical libraries that have been created may enable a weak perturbing agent to be identified, which can then be further refined to become more potent and selective. New chemical approaches such as dynamic chemical libraries provide even greater access to unexplored chemical space. Natural products, which have been refined by evolution, also provide bioactive skeletons that can be probed for these and other new uses.
Tools for Searching for Future Drugs
Clearly, computational strategies will continue to drive the development of more powerful computers, programs, and functional databases with new targets. Public and private efforts, aspiring to determine the structures of all of the major disease-associated proteins and to produce new predictive software, are paving the way for such ongoing innovation. More productive screening, on the order of millions of compounds in a single day, has been made possible by the availability of automated protein production facilities, liquid handling systems, and detectors. Improvements in microscopic platforms, biosensors, and fluorescent probes now permit multiparametric analyses in cells and some simple organisms, enabling several simultaneous events to be examined. These tools can be applied as a primary method to identify new agents or as secondary assays to confirm the desired biological activity. Genetic methods allow investigators to delete, mutate, or express genes in cells thought to be associated with human diseases. Genetically altered bacteria, yeast, and rodents can also be employed for evaluation of compounds. Small interference RNA strategies are available to assist in the deconvolution of compounds with interesting activities (12). These developments have culminated in an integrated approach to discovery, sometimes termed a Systems Biology approach, which perhaps represents an ongoing convergence in the use of reverse and forward pharmacology.
The Drugs of the Future
So what will the drugs of the future look like? Speculation of this type is always dangerous. Nevertheless, small molecules seem likely to be dominant therapeutics for the foreseeable future. Orally active drugs will command a high premium due to ease of administration and convenience. Compounds that are difficult to formulate may benefit from advances in material sciences and nanotechnology. New delivery systems that improve absorption, sense drug or biomarker blood concentrations, and optimize release also seem likely to emerge as transformational approaches. This should allow increased use of large molecular entities, such as therapeutic proteins, antibodies, oligosaccharides, and nucleic acids. In any event, it is certain that during ASPET’s second century, there will be no shortage of opportunities for creative scientists to continue the great story that we’ve seen written thus far on the pages of drug discovery history.
Acknowledgments
The author thanks Robert J. Tomko Jr. and Pierre Edouard Queiroz-Oliveira for their thoughtful comments.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

John S. Lazo, PhD, is the Allegheny Foundation Professor of Pharmacology and the Director of the University of Pittsburgh Drug Discovery Institute. E-mail lazo{at}pitt.edu; fax 412-648-9009.

