An Old Dog Learns a New Trick: Regulation of Peripheral Glucose Homeostasis by the Serotonin (5-HT)2C Receptor
The American Diabetes Association has determined that diabetes costs the United States more than $170 billion per year in associated health care costs (1). Even more discouraging is the fact that the percentage of individuals diagnosed with type II diabetes continues to climb both in the U.S. and worldwide. Although numerous medications are prescribed to treat hyperglycemia, finding effective treatments for diabetes and the resulting microvascular-related complications remains a challenge. Thus discovery of new modalities by which to treat this condition is of the utmost importance.
Diabetes begins with a loss of insulin sensitivity in white adipose tissue and skeletal muscle. Initially, the body compensates for the decrease in insulin-mediated signaling by enlarging the beta cells (found in the Islets of Langerhans in the pancreas), resulting in increased insulin release to maintain euglycemia (2). Eventually, however, the beta cells begin to fail, leading to a loss of insulin production. The lack of insulin results in elevated plasma glucose concentrations, which if sustained, leads to damage of the microvasculature of various internal organs (e.g., kidneys and eyes) and the peripheral aspects of the limbs, including the feet.
Current therapies for diabetes include agents that enhance insulin sensitivity, such as metformin and the thiazolindinediones; elevators of insulin release, such as the sulfonylureas and metiglinide; and inhibitors of carbohydrate absorption, such as acarbose (3). More recently characterized agents mimic the role (and enhance the activity) of the incretin hormone glucagon-like peptide-1 (GLP-1) (Box 1). Such mimetics include the dipeptylpeptidase inhibitor sitigliptin, which prolongs the half-life of endogenous GLP-1, and the non-hydrolyzable GLP-1 analog exendin-4. GLP-1 is released in a glucose-dependent manner from the L cells of the gut, enters the circulation, evokes GLP-1 receptor–mediated release of insulin from beta cells, inhibits glucagon release, and reduces gastric motility (4).
Incretin Mimetics
“Incretin mimetics are a new class of pharmacological agents with multiple antihyperglycemic actions that mimic some effects of endogenous incretin hormones, including glucose-dependent enhancement of insulin secretion. Although these agents may exhibit glucoregulatory effects similar to those of glucagon-like peptide-1 (GLP-1), their actions might not be mediated solely through the pancreatic GLP-1 receptor. Therefore, the class name ‘incretin mimetic’ is intended to emphasize the glucoregulatory and metabolic effects of these agents, rather than their specific mechanisms of action.” From (42).
Serotonin (5-HT)-mediated neural networks—and the 5-HT2C receptor (5-HT2CR) in particular—have long been implicated in the control of food intake and energy homeostasis. Evidence for this comes from both pharmacological and genetic experiments. Fenfluramine, a stimulator of serotonin release and an inhibitor of serotonin reuptake, was marketed as a weight loss agent from 1973 until 1997 (5), when it and its more potent enantiomer dexfenfluramine were removed from the market because of their cardiac side effects, which were likely due to activity at the 5-HT2BR (6). These weight loss effects are 5-HT2CR–mediated, as is demonstrated by the reduction in efficacy of m-chlorophenylpiperzine (mCPP) and fenfluramine when administered to 5-HT2C knockout (KO) mice (7, 8). Thus, highly selective 5-HT2C receptor agonists have been extensively studied over the last decade as a drug development target for obesity and more recently as a possible treatment for schizophrenia. Two compounds, lorcaserin and vabicaserin, developed for the treatment of obesity and schizophrenia, respectively, have entered late-stage trials and might reach the marketplace in the next two to five years. New research by Zhou et al. implies that these and other 5-HT2CR agonists may have utility in the treatment of type II diabetes (9). If true, these findings would indicate that a distinctly neuronal mechanism may be an effective treatment option for regulating plasma glucose concentrations.
Zhou et al. identify 5-HT2CR agonists as possibly useful agents for type II diabetes and demonstrate that the action of these compounds is dependent upon the melanocortin-4 receptor (MC4R) system (Figure 1⇓). The hypothesis that 5-HT2CR agonists may improve glucose tolerance was based on the finding that the 5-HT2CR KO mouse develops hyperinsulinemia, type II diabetes, and obesity as it ages. The authors utilized the 5-HT2C agonist mCPP and a presumably more selective proprietary agonist from Biovitrum, termed BVT.X, for their studies. The authors tried to dissociate lowering of plasma glucose concentrations from effects on food intake by using sub-anorectic doses of each molecule. After fourteen days of treatment with mCPP, diet-induced obesity (DIO) mice exhibited reduced circulating insulin concentrations while blood glucose, body weight, and feeding remained unchanged. Fourteen days of BVT.X treatment in ob/ob mice produced similar findings. In hyperinsulinemic DIO mice, a single dose of mCPP improved glucose tolerance as measured by an oral glucose-tolerance test (OGTT). The effect was blocked by RS-102221, a selective and brain-penetrating 5-HT2CR antagonist. A second OGTT in DIO mice revealed that reduction in plasma glucose concentrations could be blocked by intra-cerebroventricular (icv) administration of SHU9119, an antagonist of MC3Rs and MC4Rs. Further testing, however, determined that the effect of mCPP was still present in MC3R KO mice but absent in mice lacking the MC4R, indicating that the melanocortin system, working specifically through MC4Rs, is required for 5-HT2CR-mediated effect on glucose homeostasis. Indeed, members of this same team have previously demonstrated that the anorectic effect of 5-HT2C activation is melanocortin-dependent and that the 5-HT2CR is located on proopiomelanocortin (POMC) neurons of the arcuate nucleus of the hypothalamus (10). Unfortunately, no dose-response analyses using mCPP in the OGTT assays were conducted in this article. As all the acute studies were performed in overnight-fasted animals, it should have been possible to test higher doses of the compound as food intake reductions would not confound the OGTT results. These studies would have been informative insofar as the maximal efficacy of the 5-HT2C system on glucose control could have been determined. Also of relevance is a recent paper on lorcaserin that reports 5-HT2CR-mediated induction of penile erection in rats (which might also be mediated by melanocortin signaling) exhibits a bell-shaped dose–response curve (11), and so it cannot be ruled out that the same phenomenon could be observed for glucose regulation as well.
mCPP is the most often used and widely available 5-HT2C agonist; however, the compound has potent activities at many 5-HTRs in vitro (including the anorectic 5-HT1BR) and exerts behavioral effects in 5-HT2CR KO mice (12). The authors addressed this concern by using an apparently more selective agonist in BVT.X, a compound whose affinity for several 5-HTR subtypes appears to be very selective (13). The recent commercial availability of somewhat more selective 5-HT2C agonists such as WAY-161503, VER-3323, and Ro-60175 will aid further research into the role of the receptor on glucose regulation.
Although the Zhou et al. paper presents novel, compelling evidence of serotonergic pathway involvement in diabetes and glucose homeostasis, indications that these systems are involved in glucose homeostasis have appeared in the past. As with the link between 5-HT2C and body-weight regulation, these hints have come from both pharmacological and genetic experiments. In human diabetics, fenfluramine improves glucose tolerance and insulin action (14), and dexfenfluramine improves glucose control in rat models of diabetes (15). Further evidence for a role of the central serotonin system in glucoregulation comes from the finding that certain antipsychotics, which are at least partial 5HT2CR antagonists, can cause hyperglycemia (16). Antidepressants, on the other hand, have been associated with bidirectional effects on blood glucose, depending upon whether they act at the 5HT2CR (hyperglycemia) or at the serotonin reuptake transporter (hypoglycemia) (17). Studies designed to directly test the effects of 5HT2CR agonists on diabetes end-points support a role for this receptor in glucoregulation. Fenfluramine (14) and dexfenfluramine (18) improve glucose tolerance, as measured by OGTT, in patients with type II diabetes. As fenfluramine exerts much of its biological action via its metabolite norfenfluramine (19), a known potent 5-HT2CR agonist, it is perhaps not surprising that the direct-acting 5-HT2CR agonists had similar actions in mice.
Further evidence for the role of 5HT2C in glucose homeostasis comes from the 5HT2CR KO mouse, first produced by Tecott’s group in 1995 (7, 20). Mice harboring genetic deletions of 5HT2CR are resistant to the anorectic effects of fenfluramine (8). These 5HT2CR KO mice are hyperphagic, and become obese by six months of age. This obesity is accompanied by hyperinsulinemia, and impaired glucose tolerance (20). Given that these effects are seen sooner, and become more pronounced when the mice are maintained on a high fat diet, diabetes was thought to be a consequence of obesity (21). This potentially false conclusion brings up one of the most important points of the Zhou paper (9). The effects of 5HT2CR agonists on glucoregulation have traditionally been difficult to tease apart from the weight-reducing effects of these drugs—weight loss alone is sufficient to improve blood glucose control and insulin sensitivity, and many of the clinical studies showing positive glucose effects with 5HT2CR antagonists were conducted with patients on long-term treatment. Zhou and colleagues clearly demonstrate that mCPP had effects on blood glucose at doses below those needed to see effects on food intake or body weight, indicating a weight loss–independent role for the 5HT2CR in glucose regulation.
It should be noted that the melanocortin system has also previously been implicated in glucose homeostasis. The MC4R KO mouse develops dramatic obesity and hyperglycemia (22). Fan et al. report that the MC4R agonist melanotan-II (MTII), administered centrally, increased blood glucose concentrations and worsened glucose tolerance in an intraperitoneal GTT (23). In the same paper, the authors demonstrated that the MC4R KO mice exhibited impaired insulin sensitivity and hyperglycemia before the onset of its phenotypic hyperphagia and obesity. The authors of the Zhou paper propose that the effects of the melanocortin system on peripheral glucose may be downstream from those of the 5HT2CR. Strengthening this conclusion is a recent report indicating that the large majority of the POMC neurons in the arcuate nucleus have 5HT2CRs and are stimulated by 5HT2CR agonists (13).
So how can a receptor exclusively localized to the CNS modulate glucose and insulin levels in the periphery? In the current study Zhou and colleagues were able to determine that 5-HT2C stimulation activated c-fos expression in the intermediolateral nucleus of the spinal cord, which expresses MC4Rs, and that the c-Fos protein expression was lost in the MC4R KO animals (9). The intermediolateral nucleus of the spinal cord (IML) is part of the system that relays hypothalamic signals to the sympathetic nervous system, via the brainstem, to peripheral regions including brown adipose tissue and the liver in mice. Activation of the sympathetic system was then concluded to be responsible for the acute mCPP-induced enhancement of protein kinase B (PKB) phosphorylation in the liver and skeletal muscle.
Peripheral glucose homeostasis is thought to be centrally mediated; however, only recently have the neurochemical pathways underlying such regulation begun to be identified. For a central regulation of peripheral glucose homeostasis to occur, the brain must be able to: 1) determine the energy state of the body, and 2) effect at change in that state through behavior and metabolic shift. Note that nutrient detection may be indirect, as in the detection of nutrient-induced peripheral signals (e.g., insulin or leptin), or direct, such as the detection of glucose or fatty acids via biochemical-sensing cells.
Recently, the hypothalamus, and more specifically the arcuate nucleus, has been implicated as the primary nutrient-sensing site in the brain (24, 25). The central application of nutrients known to reduce feeding, such as glucose or fatty (oleic) acids, also decreases circulating blood-glucose concentrations, indicating central sensing of the nutrient molecules (26–28). Insulin supplied directly to the arcuate nucleus suppresses hepatic glucose production (26). This pathway would appear to be melanocortin mediated. Insulin delivered to the brain ventricles increases expression of POMC and reduces food intake. This effect is blocked by the MC4R antagonist SHU9119 (29). During insulin clamp studies, the rate of glucose infusion was markedly increased by α-melanocyte stimulating hormone (α-MSH) and markedly decreased by SHU9119 (30). Central inhibition of fat oxidation results in elevated peripheral glucose production, demonstrating that the brain is sensing fatty acid. This elevation in peripheral glucose production is vagally mediated, as cutting the descending hepatic vagus abolishes this effect (28).
The molecular signaling pathways mediating the effects of these nutrient-sensing neurons are also being elucidated. Of interest for the present discussion is AMP-activated protein kinsae (AMPK). The mammalian target of rapamycin (mTOR) may also be of interest, but is outside of the scope of the present commentary. AMPK is found in various tissues throughout the body, and acts as a switch between anabolic and catabolic states. Specifically, AMPK is activated when ATP is depleted (31). This molecular sensor also seems to be influenced by melanocortin tone. Minokoshi et al. demonstrated that leptin, insulin, MT-II, and glucose decrease AMPK activity in the hypothalamus (32), whereas Claret et al. have used elegant genetic techniques, ablating AMPK in selective neuronal populations, to demonstrate that AMPK is essential for the activity of agouti-related peptide (AGRP)- and POMC-releasing neurons to sense glucose and alter energy homeostasis (33).
Together these results begin to illustrate how a brain senses nutrients at the level of the hypothalamus. This nutrient sensing can be direct or hormonally mediated. The result of this sensing includes regulation of circulating peripheral glucose, and in many cases this process is influenced by the central melanocortin system (34). This sensing may activate hindbrain areas associated with vagal nerve activity (35): transection of the descending (efferent) vagus can reduce the peripheral metabolic effects of central nutrient manipulation (27, 35, 36).
The importance of hindbrain areas and efferent vagal signals in these effects is interesting when considering the paper under discussion. Although it has been assumed that the glucose-regulatory effects of 5HT2CR agonists are regulated by hypothalamic 5HT2CRs, Grill and colleagues (37–39) have elegantly demonstrated that the 5HT2CR agonists and MC4R agonists and antagonists can alter food intake and energy homeostasis when their effects are restricted to the hindbrain alone. Given the proximity of these hindbrain receptors to the major influx and outflow of neuronal activity from the vagus nerve and the importance of vagal efferent neurons in the metabolic effects of central nutrient detection, the relative role of efferent vagal signals in the 5HT2CR–melanocortin mediation of blood glucose concentrations should be addressed.
One other aspect of this work that may be of importance relates to the pro-diabetic actions of some atypical antipsychotic drugs as some of these agents, including olanzapine, are antagonists of the 5-HT2CR. Indeed 5-HT2CR antagonism has previously been implicated in the weight-promoting action of these drugs, and now one must consider whether disregulation of glucose homeostasis may also be a consequence of 5-HT2CR blockade (40). In contrast, if selective 5-HT2CR agonists indeed prove to be effective in treating schizophrenia, an added benefit of weight loss and improvement in glucose control may be observed as schizophrenics tend to present with metabolic disregulation at a rate higher than the general public (41).
We will soon know if selective 5-HT2CR agonists can reduce glucose concentrations in humans to the same degree as fenfluramine. Arena Pharmaceuticals have announced they will commence a Phase III study—Behavioral Modification and Lorcaserin for Overweight and Obesity Management in Diabetes Mellitus (BLOOM-DM)— with their selective 5-HT2C agonist lorcaserin in patients with Type II diabetes. If successful, 5-HT2CR agonists may signify new, successful molecular interventions by which to treat significant elements of the metabolic syndrome.
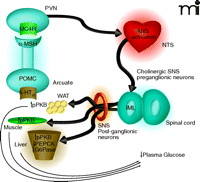
Proposed mechanism by which 5-HT2C receptor agonists regulate peripheral glucose. Activation of 5-HT2C receptors (5-HT2CRs) expressed on proopiomelanocortin (POMC) neurons of the arcuate would lead to elevated α-MSH release in the periventricular nucleus (PVN). Activation of MC4 receptors (MC4Rs) in the PVN would be followed by downstream activation of the autonomic nervous system (ANS), likely in the region of the nucleus of the solitary tract (NTS). Based on the findings by Zhou et al. (9) of c-Fos expression and co-localization with choline acetyltransferase (ChAT), the intermediolateral nucleus (IML) is likely a relay point between the CNS and periphery. As the IML is part of the sympathetic nervous system (SNS) one would expect that post-ganglionic neurons of the SNS are then activated to effect changes in the muscle, white adipose tissue (WAT) and liver that influence glucose production, sensitivity, or both. The increased level of protein kinase B phosphorylation (pPKB) observed in the three tissues is suggestive of improved insulin sensitivity. The 5-HT2C-mediated reductions in phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6P) mRNA in the liver are classically associated with enhanced insulin signaling in that organ and would be expected to result in reduced gluconeogenesis.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Anthony V. Azzara, PhD, obtained his degree at the City University of New York, investigating neural and behavioral mechanisms underlying food-based learning. His post-doctoral work was conducted at the E.W. Bourne Behavioral Research Laboratory at the Weill Medical College of Cornell University. There he studied the role of gut-brain communication in the control of feeding behavior. Subsequently he joined the faculty of the Albert Einstein College of Medicine in the Department of Endocrinology, continuing to study food intake and energy homeostasis. He is now a Senior Research Investigator in Discovery Biology at Bristol-Myers Squibb, focusing on the obesity therapeutic area.

Keith J. Miller, PhD, obtained his doctoral degree at the Albany Medical College, characterizing the pharmacology of various serotonin receptors. His post-doctoral work was conducted in the Laboratory of Cell Biology at the NIMH in Bethesda, MD. There he studied the regulation of neurotransmitter transporters by nitric oxide and other second messengers. He then joined the Nova Southeastern University College of Pharmacy where he studied interactions between the serotonin 5-HT2A receptor and nitric oxide synthases. He is now a Senior Principal Investigator at Bristol-Myers Squibb, focusing on G protein–coupled receptors involved in metabolic disease. E-mail Keith.Miller{at}bms.com; fax 609-818-3239

