G Protein-Coupled Receptors Go Extracellular: RhoA Integrates the Integrins
Abstract
The identification of downstream effectors of G protein–coupled receptors (GPCRs) is critical for understanding the interactions between signaling cascades and for developing new pharmacological approaches for controlling GPCR-mediated responses. RhoA is a small G protein that serves as a proximal downstream effector of numerous GPCRs and regulates a variety of basic cell functions, including migration, survival, and proliferation. Intriguingly, GPCR ligands such as thrombin, sphingosine-1-phosphate, and lysophosphatidic acid, which signal through G12/13 and activate RhoA, have recently been shown to induce the expression of the extracellular matrix protein Cyr61 (i.e., CCN1). Cyr61 is secreted and interacts with cell surface integrins to activate kinase and transcriptional cascades that are also known to contribute to cell migration, survival, and proliferation. The GPCR/RhoA/Cyr61/integrin pathway defines a novel convergence mechanism for integrating GPCR-and integrin-dependent signaling cascades that may contribute to sustained and pathophysiological responses to GPCR activation.

Introduction
G protein–coupled receptors (GPCRs) span the cell membrane and transduce extracellular messages from soluble ligands binding at the cell surface into intracellular second messengers. These messengers initiate signaling cascades that ultimately control myriad cell responses. Proteins in the extracellular matrix, acting through integrin receptors on the cell surface, also transduce messages that elicit cell responses. The relationship between GPCR and integrin pathways has not been well explored. This review presents data suggesting that activation of GPCRs induces the expression of Cyr61 (i.e., CCN1), a protein which, upon secretion, activates integrins, thereby providing signals that complement and contribute to GPCR-mediated responses. Interestingly, both the GPCR and integrin pathways require the small G protein RhoA for appropriate signal transduction, suggesting a further interrelationship between the extracellular matrix and a signaling via a subset of GPCRs.
The Extracellular Matrix, Integrins, and RhoA
The extracellular matrix (ECM) is composed of proteoglycans, glycoproteins, and complex polysaccharides that interact to provide a structural scaffold and informational cues. The composition of the ECM can change as a function of the “remodeling” processes that occur during thrombosis and wound healing, differentiation, and a wide variety of pathological processes. Notably, local remodeling of the ECM can occur quickly during these processes, as the induction of new ECM proteins is dynamically influenced by cues from the local microenvironment.
The principal receptors for the ECM are integrins, heterodimeric proteins composed of at least one beta and one alpha subunit. From among the cellular repertoire of eighteen alpha subunits and eight beta subunits, at least twenty-four distinct heterodimers can be formed, each binding to distinct ECM ligands. Unlike classical growth factor receptors, integrins bind ligands in a mechanosensory fashion that requires the assembly of a “focal adhesion complex” associated with the actin cytoskeleton. Assembly of a mature signaling complex requires integrin ligands to provide mechanical resistance, which opposes the contractile force of the associated actin cytoskeleton and promotes continued ligation events as new integrins are recruited into the growing focal complex. Smaller soluble ligands, such as those released from or produced by proteolytic modification of the ECM, can prevent positive signaling as integrins become saturated with soluble rather than immobilized ligands. However, at lower concentrations, these soluble ligands promote signaling by facilitating integrin cycling between ECM-ligated and unligated states.
Typically, extracellular proteins such as collagen, fibronectin, and laminin are tightly associated with the ECM. In contrast, the class of proteins termed “matricellular” can associate more loosely with the ECM, fulfilling roles as soluble or ECM-anchored ligands. These proteins are often upregulated during matrix remodeling and differentiation processes, and they appear to promote signals that facilitate cell proliferation and migration, at least in part via integrins.
Cysteine-rich protein 61 (Cyr61/CCN1), first identified as a growth factor–inducible immediate early gene, is one of the prototypical members of the CCN family of matricellular proteins. This family includes the well-studied connective tissue growth factor (CTGF/CCN2) and additional proteins (Figure 1⇓) that differ in their regulation, tissue distribution, and biological function. Early studies by the Lau laboratory demonstrated the importance of Cyr61/CCN1 in fibroblast attachment, spreading, and chemotaxis (1). In addition, although not a mitogen in its own right, Cyr61/CCN1 promotes proliferative responses to growth factors through its interaction with cell surface integrins (1–3).
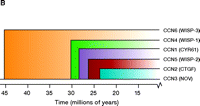

The CCN family of proteins. A modular representation of the human CCN1 protein indicates i) the signal sequence; ii) the insulin-like growth factor–binding protein homology domain (IGFB, blue); iii) the von Willebrand factor type C domain (VWC, green); iv) the thrombospondin type 1 homology domain (TSP1, orange); and the cysteine knot–containing C-terminal domain (yellow). Semi-transparent colored bands around the domains indicate sites that interact with integrins (e.g., integrin αvβ3, α6β1, and αMβ2); also shown are the two binding sites for heparin (purple C-terminal bands). The dashed line indicates the hinge region; numbers represent amino acid positions in the protein sequence. The dendrogram reflects sequence conservation among the human CCN proteins (20). See text for details.
Integrin signaling depends on the effects of RhoA on the actin cytoskeleton, and interestingly, integrin ligation also activates RhoA (4). As evidence of interactions between integrin and RhoA pathways, fibroblasts from mice lacking focal adhesion kinase (FAK) compensate for this deficiency in part via upregulation of RhoA signaling (5). Widely appreciated for its functions in controlling the actin cytoskeleton (6, 7), RhoA also serves to mediate cell proliferation and gene expression in response to GPCR ligands (8–10).
RhoA is activated via GPCRs that couple to the heterotrimeric G12/13 proteins. This response is catalyzed by guanine nucleotide exchange factors (GEFs), several of which are direct targets (effectors) of the G12/13 alpha subunits (Figure 2⇓). Ligands that activate RhoA through coupling of their receptors to G12/13 include thrombin, sphingosine-1-phosphate (S1P), lysophosphatidic acid (LPA), and thromboxane A2 (9, 11–15). These are not considered classical neurotransmitters, but rather they are mediators released or formed under conditions of inflammation and at sites of injury. All of these agents are notable for their ability to induce cell proliferation and/or migration (9, 16–18, 71). Thus, activation of these GPCRs, like engagement of integrins through Cyr61/CCN1, contributes to pathophysiological responses. The extent to which GPCR-mediated activation of RhoA is related to integrin signaling is not known. Significantly, the transcriptional regulation of Cyr61/CCN1 expression by this same set of GPCRs appears to be closely linked to RhoA activation. Thus, we have proposed Cyr61/CCN1 signaling as a likely downstream event contributing to altered cell migration or proliferation induced by these “inflammatory” GPCR ligands and involving integrin activation.
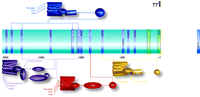
Promoter sequences from the CCN1-encoding gene and selected transcriptional response elements. The center portion of the Figure lists promoter elements that have been determined through studies of the human (white text) and murine (black text) gene. Above and below the schematized promoter are GPCR-mediated signaling pathways that could regulate expression of the gene by targeting selected promoter elements. GPCRs (colored structures of seven cylinders) are located in the plasma membrane and preferentially couple to distinct G proteins as indicated (see text for details).
The CYR61/CCN1 Protein Family
The Cyr61 gene that encodes the CCN1 protein (42 kDa) was cloned in 1990 from a serum-stimulated BALB/c 3T3 fibroblast cDNA library (19). In addition to Cyr61/CCN1, the protein family includes connective tissue growth factor (CTGF/CCN2), nephroblastoma overexpressed gene (Nov/CCN3), and the Wnt-induced secreted proteins WISP-1, -2 and -3 (WISP1–3/CCN4–6). The CCN protein family members share thirty to fifty percent overall amino acid sequence identity and forty to sixty percent sequence similarity with one another (2, 20, 21).
As indicated in Figure 1B⇑, each CCN family member possesses four distinct domains. The insulin-like growth factor–binding protein domain is fully functional and can serve as a depot for insulin-like growth factors within the ECM or at the cell surface. The von Willebrand factor type C domain and the thrombospondin I domain likely function to promote protein–protein interactions within the ECM. The C-terminal module of all CCN proteins except CCN5 includes both a heparin-binding and a cysteine-knot motif; aside from permitting association with additional integrins and the proteoglycan component of the ECM, this domain also mediates disulfide bonding between CCN proteins. Figure 1B⇑ also shows the integrin-binding regions of Cyr61/CCN1 that may mediate cell type-specific activities (22–25).
Typical for a matricellular protein, Cyr61/CCN1 interacts both with cell surface receptors and additional ECM components. Originally described as an integrin αvβ3– and heparin-binding protein (26), Cyr61/CCN1 has subsequently been shown to be promiscuous in its choice of binding partners. Cyr61/CCN1 is bound by integrins αvβ3, αvβ5, α6β1, αIIbβ2 and αMβ2 [see (27)]; the binding sites for αvβ3 (22), α6β1 (23), αMβ2 (24), and heparin (25) have been identified (see Figure 1B⇑). Processes such as adhesion (22, 23, 28–30), migration (29), growth factor–induced proliferation (29), and chemotaxis (30) have been attributed to ligation of Cyr61/CCN1 to specific integrins. Cell adhesion to Cyr61/CCN1 via α6β1 also requires the presence of heparin sulfate proteoglycans (HSPGs) (25, 30), whereas adhesion mediated by other integrins (i.e., αvβ3) does not (25).
All of these integrins bind to the RGD (Arg-Gly-Asp) recognition sequence present in adhesive proteins such as fibronectin, fibrinogen, vitronectin, and von Willebrand factor. Indeed, peptides that contain the RGD sequence can competitively inhibit the binding of these adhesive proteins to integrins [see (31)]. Interestingly, Cyr61/CCN1 does not contain an RGD sequence, yet has been shown to mediate cell adhesion through RGD-binding integrins. Accordingly, the interaction of Cyr61/CCN1 with RGD-containing integrins is blocked by RGD-peptides (27).
GPCRs and the Regulation of Cyr61/CCN1 Expression
Cyr61 (i.e., the Cyr61/CCN1-encoding gene) is expressed at low levels in most quiescent cells, but its transcription is upregulated in response to serum and growth factors. This upregulation can be sustained for many hours [see (2) and Figure 3⇓], which differentiates Cyr61 from many immediate early genes and also suggests involvement in chronic responses, possibly through positive feedback regulation. Moreover, the variety of factors that can induce the expression of this gene is remarkable. These include growth factors (PDGF, VEGF, EGF, bFGF), cytokines (IL1,2,6 and TGFβ), vitamin D3, tamoxifen, and cortisol [see (20, 32)], as well as GPCR agonists [S1P, LPA, thrombin, angiotensin II, phenylephrine, and PGF2α (33–42); also discussed below]. Indeed, if the importance of a protein can be judged according to the number of paths that lead to its upregulation, both Cyr61/CCN1 and CTGF/CCN2 rank high. Although these and other clues in the literature hint at the potential regulatory role for CCN proteins as mediators of signal transduction pathways, interest in these proteins has tended to remain primarily within the purview of investigations into the ECM.
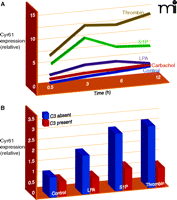
Kinetic expression profile of Cyr61/CCN1 after GPCR stimulation and Rho dependence of CCN1 protein expression after GPCR stimulation. (A) CCN1 kinetic expression profile in 1321N1 glioblastoma multiforme cells treated with vehicle (Control), 500 μM carbachol, 10 μM lysophosphatidic acid (LPA), 10 μM sphingosine-1-phosphate (S1P), or 0.5 U/ml thrombin. Mean densitometric values for CCN1 normalized by actin are shown. (B) 1321N1 cells were treated overnight with C3 toxin from Clostridium botulinum followed by stimulation with 10 μM lysophosphatidic acid (LPA), 10 μM sphingosine-1-phosphate (S1P), or 0.5 U/ml thrombin for 3 hours.
Our own attention was drawn to these CCN proteins as a result of the unique gene expression profiles that we generated by comparing responses of human glioblastoma cells to several GPCR agonists. These studies compared responses to thrombin, the mitogenic GPCR agonist for protease activated receptor-1 (PAR1), with those to carbachol, a non-mitogenic GPCR ligand for the muscarinic acetylcholine receptor (AChR). In this analysis, we found CYR61/CCN1 to be the most robustly and selectively activated gene at all times examined (43). Although thrombin and carbachol stimulate many common signaling pathways in this glioblastoma cell line, they are distinguished by their ability to activate RhoA, stimulate AP-1 gene expression, and induce DNA synthesis (15, 44–46). How RhoA activation, gene expression, and cell proliferation are connected has not been fully clarified, but Cyr61/CCN1 was an attractive candidate for a gene, induced through Rho and AP-1 signaling, that might contribute to mitogenic responses to GPCRs.
Thrombin and PAR-1 activation had been previously shown to induce CCN-1 expression in fibroblasts (33, 35) but the underlying signaling pathways were not explored in these studies. Of particular interest, however, was a paper from Chaquor’s laboratory demonstrating that Cyr61/CCN1 was regulated by the GPCR agonist S1P through a pathway that utilized Rho/Rho kinase and AP-1 signaling (39). In further analysis of pathways leading to the induction of Cyr61/CCN1 by S1P in smooth muscle cells, Han et al. (39) mapped the transcriptional response elements involved and demonstrated a requirement for both CRE and AP-1 sites. Pharmacological inhibitor studies ruled out involvement of PKA, ERK, or PKC in the response to S1P, although there was significant attenuation of the response by inhibitors of p38 or JNK. Most notably, the response was fully abolished by pharmacological inhibition of Rho kinase, an important effector of RhoA. These authors also demonstrated that heterologous expression of constitutively active RhoA, but not of small GTPases Rac or Cdc42 stimulated the Cyr61/CCN1 promoter (39). Another recent report demonstrates that activation of S1P2 receptors by S1P activates RhoA and induces the expression of Cyr61/CCN1 in glioblastoma cells (40); we also previously implicated the S1P2 receptor subtype in RhoA activation (47).
LPA elicits robust increases in CYR61/CCN1 expression in human prostatic epithelial cells (41), and although the underlying mechanisms have not been delineated, LPA is another highly efficacious activator of RhoA (11, 15, 48). Furthermore, our recent work links Cyr61/CCN1 upregulation through LPA, S1P, and PAR1 receptors to RhoA activation, demonstating nearly complete loss of these responses when Rho signaling is inhibited with the C3 toxin (Figure 3⇑) (43). Another interesting recent finding is that infection of epithelial cells with an attenuated Yersinia bacteria induces Cyr61/CCN1 expression through an LPA receptor–mediated mechanism (42). Intriguingly, a virulence plasmid inhibited this response in part through YopT, a factor that inactivates RhoA (49). Additionally, Wnt3A, acting through its canonical pathway, which utilizes the heptahelical receptor, frizzled, also induces Cyr61/CCN1 expression (50). Overall, the observation that there is robust Cyr61/CCN1 upregulation by agonists that act on receptors that couple to G12/13 and/or increase RhoA activation (11, 12, 16, 51–53) supports the hypothesis that Cyr61/CCN1 serves as a critical downstream signal for responses to inflammatory GPCR ligands that activate RhoA.
Although ligands for receptors coupling predominantly to Gi-, Gq- and Gs- stimulated pathways have been less frequently linked to Cyr61/CCN1 expression, there is some evidence for Cyr61/CCN1 regulation by cholera toxin (which activates the Gs protein) and by phorbol esters, which activate PKC, the down-stream target of Gq/phospholipase C activation (2, 20, 32) (Figure 2⇑). AngII, acting through the angiotensin type 1 (AT1) receptor, and phenylephrine (PE), which activates alpha1-adrenergic receptors generally thought to couple to Gq, have both been shown to induce Cyr61/CCN1 expression and its secretion into the culture medium of neonatal rat ventricular myocytes (38). Induction of Cyr61/CCN1 by PE or AngII was shown to be sensitive to pharmacological inhibitors of PKC and ERK and to be induced by PMA (38). In vivo pressure overload, elicited by transverse aortic constriction, also increases Cyr61/CCN1 expression in the heart (38); this intervention is thought to work in large part through Gq/11-coupled GPCR activation, thereby implicating Gq signaling in Cyr61/CCN1 upregulation. Although no direct role for Gi signaling in Cyr61/CCN1 expression has been demonstrated, the Akt/PI3kinase pathway, which is regulated through Gi, negatively regulates the forkhead transcription factor FOXO3a, which suppresses Cyr61/CCN1 expression in cultured rat vascular smooth muscle cells (54). In addition, AngII induces Cyr61/CCN1 expression in rat aortic smooth muscle cells (37, 54), and one study suggests that this induction reflects the modulation of FOXO3 by AngII (54). Further analysis of the kinetics and efficacy of Cyr61/CCN1 activation by a panel of GPCR agonists will be important to determine whether Cyr61/CCN1 expression by GPCR signaling occurs predominantly through G12/13 and RhoA signaling.
Role of Cyr61/CCN1 in Intracellular Signaling
Ectopic expression of Cyr61/CCN1 or its addition to culture medium elicits signaling via many kinase pathways, including PI3K/Akt (55, 56), ERK (56, 57), ILK (55), FAK (57), and Src (58) (Table 1⇓). Transcriptional pathways including TCF/LEF (55), NFκB (59), and AP-1 (39) are also activated in response to Cyr61/CCN1. Numerous genes have been shown to be upregulated in response to Cyr61/CCN1 and ultimately contribute to its cellular effects. The genes upregulated in this way include those that encode extracellular proteases [e.g., MMP-1, MMP-3, and uPA (57, 60)] and their inhibitors [e.g., TIMP1, PAI-1 (60)]; growth factors [e.g., VEGF-A and VEGF-C (60)]; and chemokines [e.g., IL-1β (60)] and chemokine receptors [e.g., CXCR1 and CXCR2 (58)]. Various integrins are similarly upregulated, including some with which Cyr61/CCN1 directly interacts [e.g., α5, α3, αv, and β1 integrins (55, 60, 61)]. Although it frequently acts as an autocrine factor, Cyr61/CCN1 has also been implicated in paracrine signaling. For example, Cyr61/CCN1 that is secreted into the culture medium by breast cancer cells causes stromal fibroblasts to secrete MMP1 (34, 62).
Downstream Events and Molecules Engaged by CCN1 Signaling
Secreted Cyr61/CCN1 associates with the ECM, and early studies by Lau and colleagues demonstrated the importance of Cyr61/CCN1 in fibroblast attachment, spreading, and chemotaxis (1). Other cytoskeletal responses are similarly regulated. Fibroblast adhesion to Cyr61/CCN1 increases formation of filopodia and lamellipodia as well as the formation of integrin α6β1-containing focal complexes. Cell adhesion to Cyr61/CCN1 results in activation of intracellular molecules associated with integrin signaling (e.g., focal adhesion kinase, paxillin, and Rac) and sustains ERK activation (57).
Cyr61/CCN1 has also been shown to play a role in regulating cell survival or apoptosis, depending on the cell and context. Ectopic expression of Cyr61/CCN1 in breast cancer cells increases levels of the anti-apoptotic protein XIAP, promoting cell survival (59). On the other hand, Cyr61/CCN1 can act as a tumor suppressor in human endometrial cancer cells by increasing expression of the pro-apoptotic proteins Bax, Bad, and TRAIL and favoring caspase 3 activity (63). In skin fibroblasts Cyr61/CCN1 induces apoptosis by triggering activation of Bax, independent of caspase-8 activity or de novo transcription (64).
There is a clear role for Cyr61/CCN1 in augmenting the proliferative effects of growth factors known to associate with the ECM. Although ineffective at inducing a proliferative responses alone, Cyr61/CCN1 acts as a co-mitogen, enhancing the responsiveness of HUVEC and NIH 3T3 cells to bFGF (1, 3). Cyr61/CCN1 can also displace bFGF from the ECM, suggesting another subtle way that Cyr61/CCN1 could modulate growth factor signaling (3). The role for Cyr61/CCN1 in cell proliferation is supported by studies examining the consequences of Cyr61/CCN1 knockdown. In VSMC, knockdown of Cyr61/CCN1 via siRNA significantly inhibits DNA synthesis in cells grown in conditioned medium (65). In our studies using astroglioma cells, thrombin-stimulated DNA synthesis is reduced by more than fifty percent following Cyr61/CCN1 knockdown (43). In addition, treatment of cells with heparin or anti-Cyr61 antibodies to bind extracellular Cyr61 (Figure 4⇓) dramatically decreases thrombin-stimulated cell proliferation (43). We suggest that this reflects the contribution of Cyr61/CCN1 and integrin signaling to the sustained cellular responses required for cell proliferation (Figure 4⇓, inset). The finding that knockdown of Cyr61/CCN1 significantly suppresses neointimal hyperplasia in a rat carotid artery balloon injury mode (65) is of further interest, as it is consistent with the involvement of GPCRs and Rho-activated pathways in vascular injury (66). Cyr61/CCN1 may thus serve as a critical mediator of proliferative and sustained responses to GPGR activation by inflammatory mediators in vivo (Figure 4⇓).
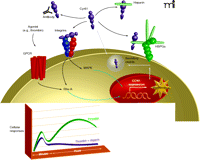
Integration of intracellular and extracellular signaling responses to GPCR activation. The schematic suggests mechanisms through which GPCRs may engage integrin signaling pathways and highlights a role for Cyr61/CCN1 in amplifying GPCR-mediated signals. Four possible fates are schematized for secreted Cyr61/CCN1: association with HSPGs in the ECM; association with heparin; neutralization by antibody; and association with integrins. The inset shows a theoretical time course, highlighting the Cyr61/CCN1-dependent (heparin-sensitive) contribution to intracellular signaling. The heparin-sensitive component is the difference between the green and purple lines.
Conclusions
Paradigms for crosstalk between signaling pathways initiated by GPCRs and those initiated by receptor tyrosine kinases are well-established in the literature. Here, we suggest the concept that engagement of the integrin pathway may likewise provide a means to amplify signals initiated through GPCRs. This concept is consistent with the long-recognized requirement for an additional stimulus to prime and maintain the signaling capacity of integrins. The discovery of a potential mediator of such crosstalk, in the form of the matricellular protein Cyr61/CCN1, provides a new way to understand how the effects of integrin and GPCR activation can be mutually complementary. Identification of Cyr61/CCN1 as a downstream effector of responses to GPCRs that activate RhoA, taken with the requirement of RhoA for appropriate integrin action, may further inform our understanding of the role of this small G protein in normal and pathophysiological cellular responses.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Joan Heller Brown, PhD, is Professor and Chair of the Department of Pharmacology at the University of California San Diego School of Medicine. Her longstanding research interest is in G protein-coupled receptor regulation of heart and astroglial cell growth and survival. Send correspondence to JHB. E-mail jhbrown{at}ucsd.edu; fax 858-822-0041.

Dwayne Stupack, PhD, is Assistant Professor in the Department of Pathology at the UCSD School of Medicine and the Moores UCSD Cancer Center. His laboratory studies the roles of integrins in cell invasion.

Colin T. Walsh, PhD, completed his doctoral work at The Ohio State University and is currently a Postdoctoral Fellow in the Heller Brown laboratory at University of California San Diego. His scientific interest is in preclinical development and clinical translation of new therapeutics for oncology.

